The migration and consolidation of legacy methods to the Waters ACQUITY UPLC System is a major focus for companies adopting UltraPerformance LC (UPLC) technology. Those companies that invest in the process are rewarded with significant savings in analysis times and operational costs while conserving or improving overall chromatographic performance.
Many laboratories are choosing to redevelop legacy methods during this migration in order to take full advantage of the strengths of the ACQUITY UPLC System, creating even more robust methods in the process. However, some legacy methods may not require full redevelopment; users can still realize significant method improvement by simply transferring their method from HPLC to UPLC.
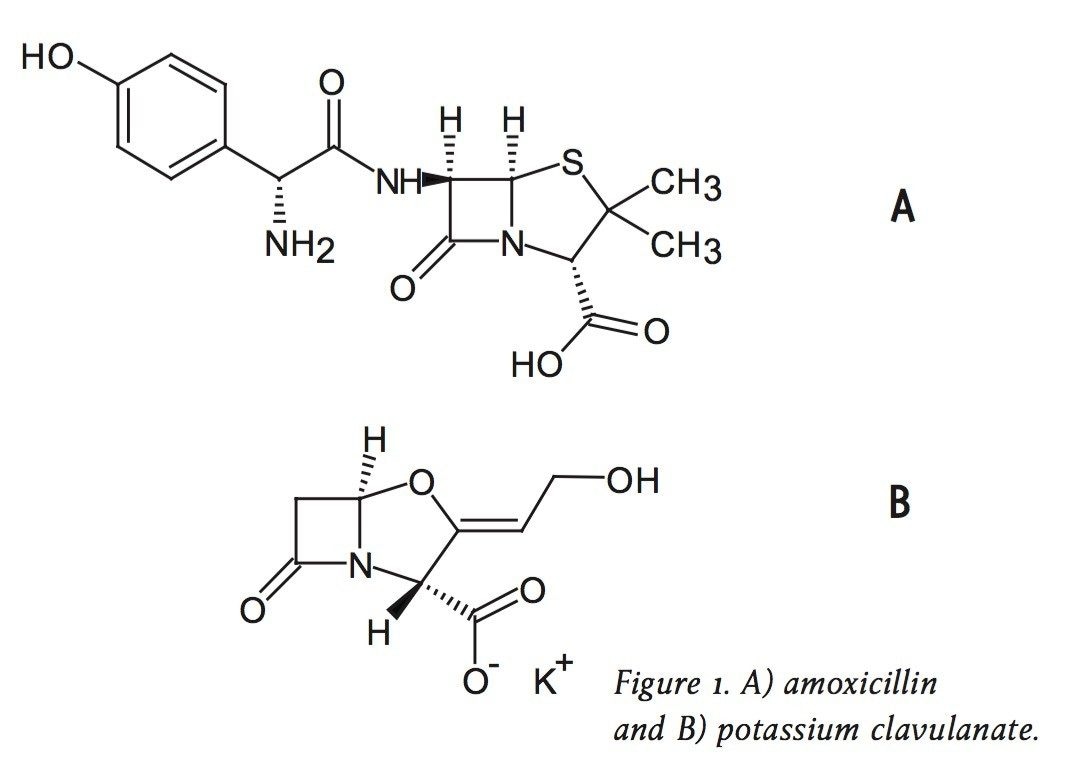
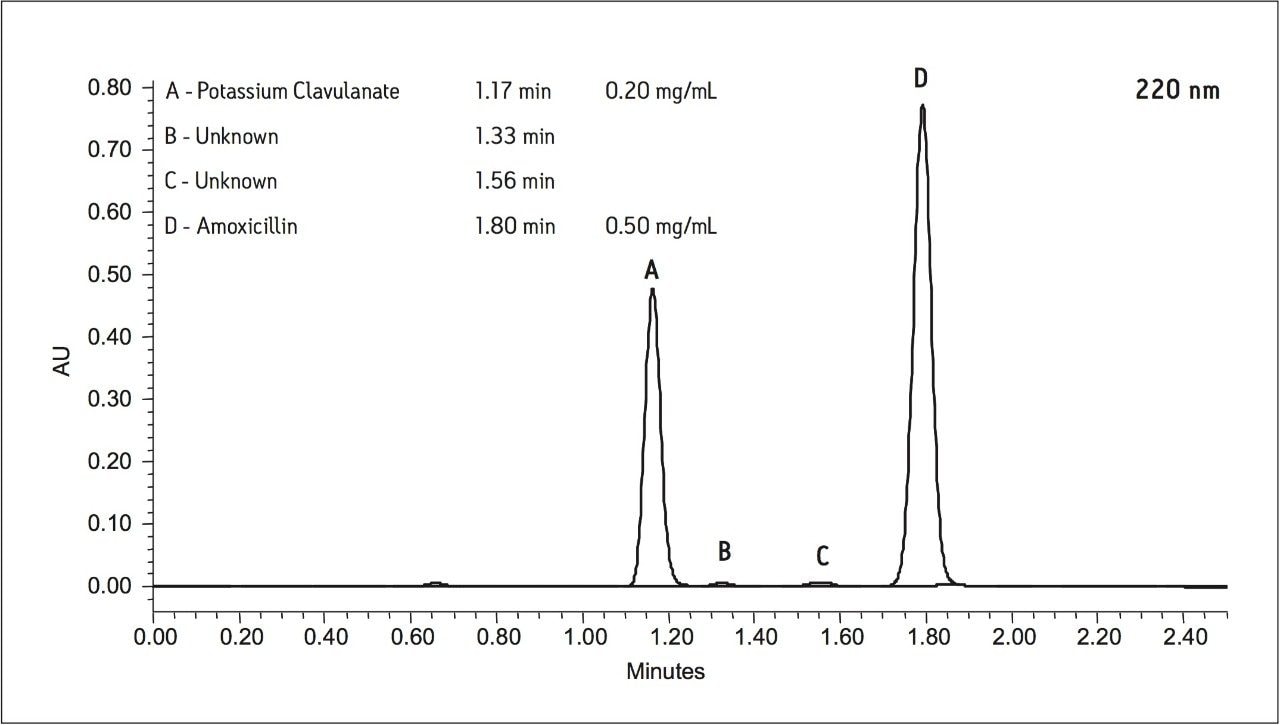
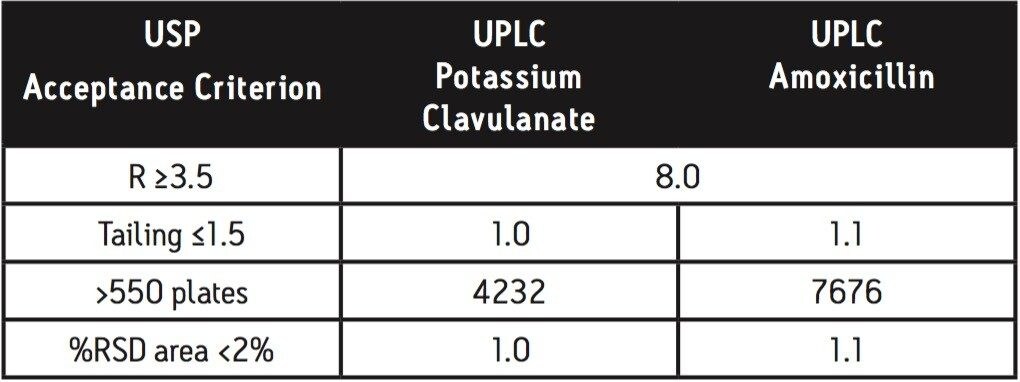
For example, the assay of amoxicillin and potassium clavulanate from an oral suspension1 is an established HPLC method that can be simply migrated to the ACQUITY UPLC with minimum time and effort. Amoxicillin (Figure 1A) is a ß-lactam antibiotic with primary activity against gram-positive bacteria. To broaden the effect of this drug on select gram-negative bacteria and to combat resistant strains, potassium clavulanate (Figure 1B) is added to formulations of amoxicillin to inhibit amoxicillin degradation by the enzyme ß-lactamase.