Peptide mass fingerprinting (PMF) by MALDI Tof mass spectrometry is a rapid and sensitive method for the identification of proteins from organisms with well characterized genomes. PMF compares experimentally observed peptide masses, obtained from the MALDI MS of enzymatically digested proteins, with theoretical peptide masses, obtained from the in silico digestion of proteins contained within protein sequence databanks. This is a highly specific, sensitive technique for identifying proteins from a known proteome. However, the PMF approach will fail when a protein is not represented in the protein sequence database (e.g. if the genome has not been sequenced). In addition, problems occur due to non-specific enzyme activity, if some of the peptides are modified or too few tryptic peptides are observed to give an unambiguous answer. In these cases, further analysis and added specificity is gained by performing MS/MS on some of the peptide molecular ions and searching this information against a sequence database. If this fails to give an unambiguous answer then homology-based searching of the amino acid sequence information may allow identification of the parent protein.
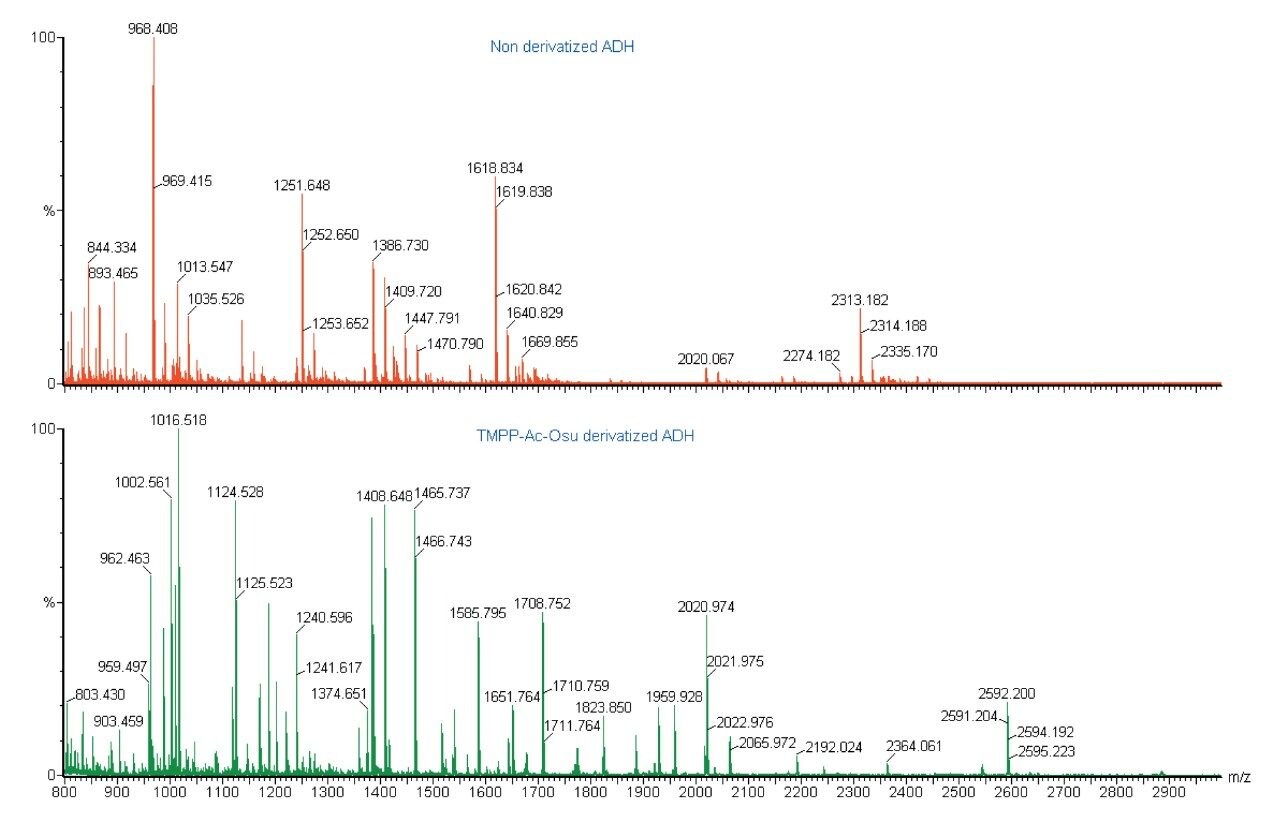
With a hybrid quadrupole orthogonal acceleration time-of-flight (Q-Tof) mass spectrometer, equipped with a MALDI source, high quality MS/MS spectra can quickly be generated from MALDI phase samples. A feature of the oa-Tof mass analyzer is the high mass accuracy that is routinely achieved—typically better than 10 ppm RMS when using a single-point lockmass. Despite the quality of the MS/MS data obtained from this geometry of instrument, the MS/MS spectra generated from singly charged peptides, produced by the MALDI ionization process, exhibit a wide variety of fragment ions. The types of fragment ions produced are very sequence dependent. This can make interpretation of MALDI MS/MS data difficult due to their complexity.
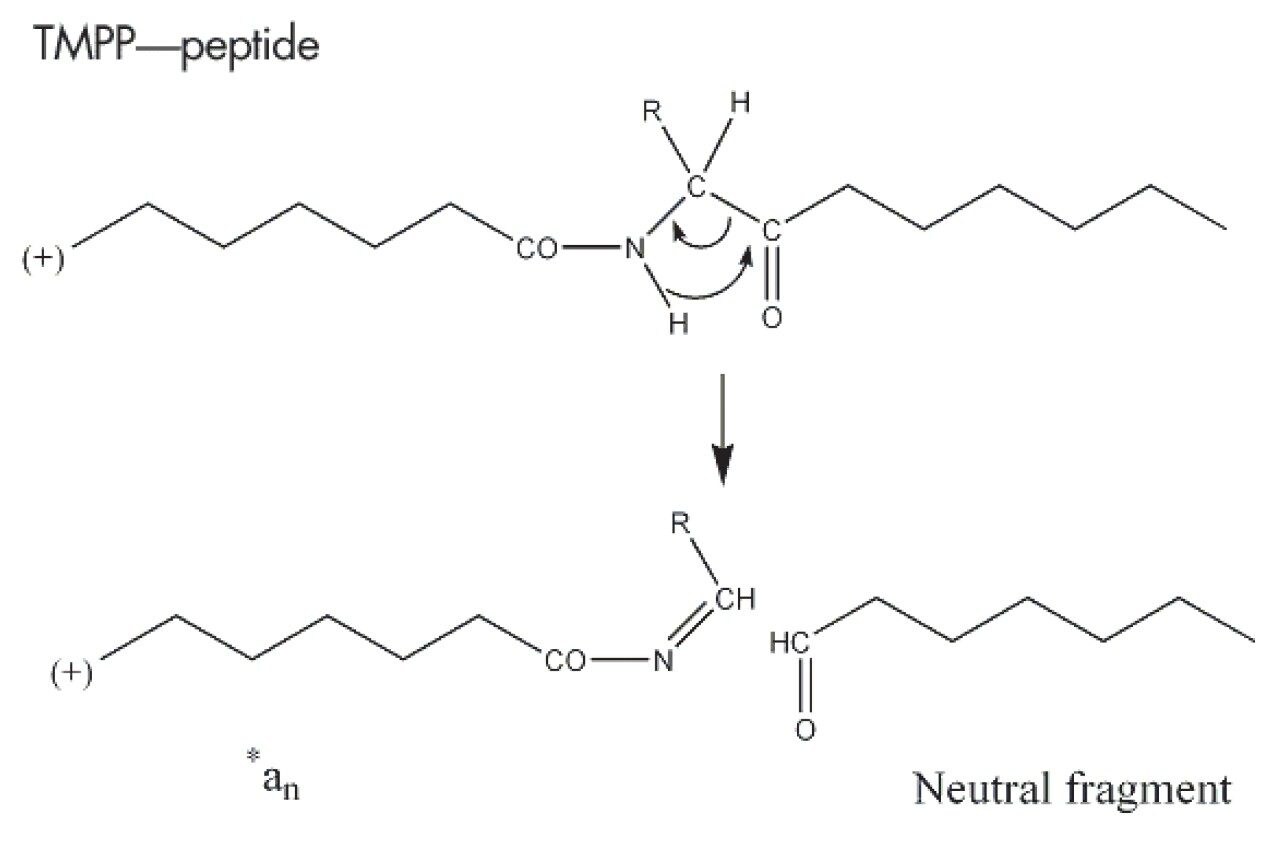
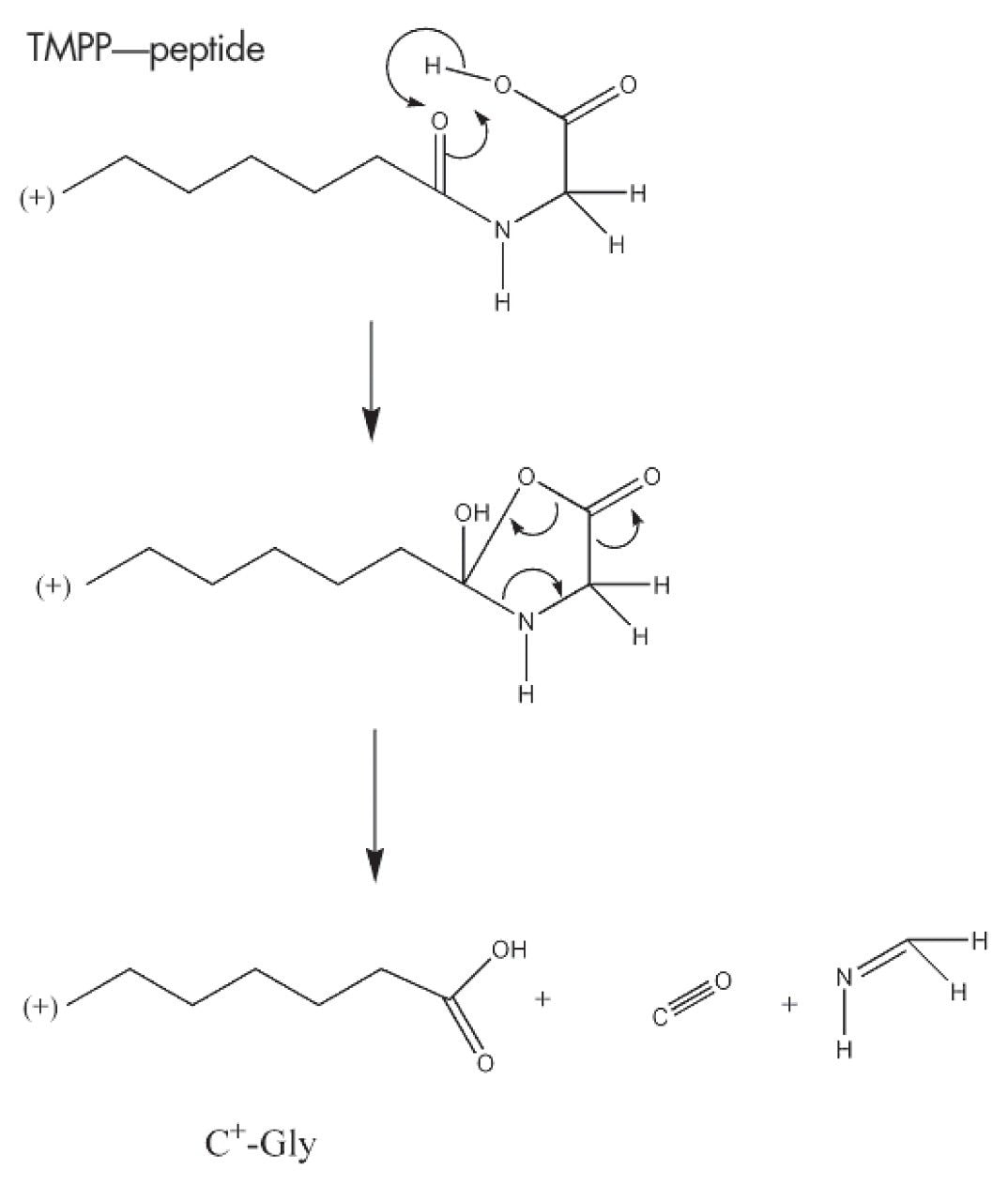
A way of simplifying the spectrum and improving the fragmentation efficiency is to drive the fragmentation process by introducing a fixed charge at a specific location on the peptide.
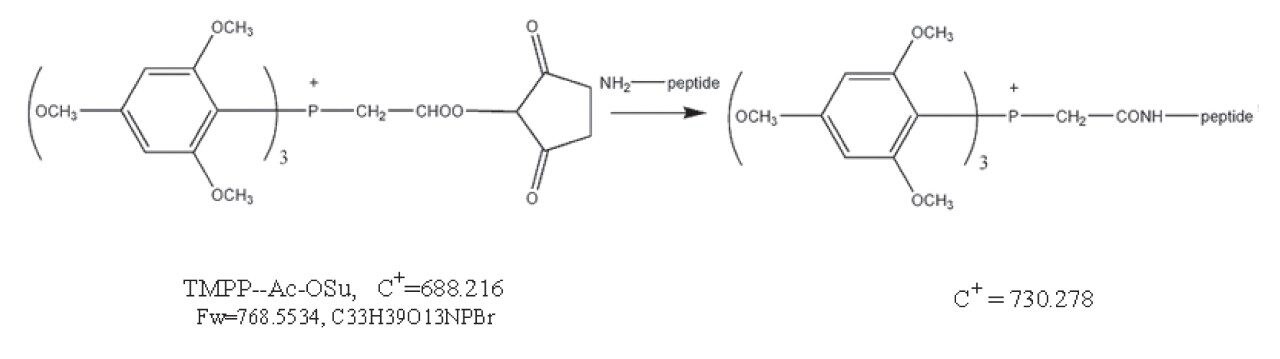
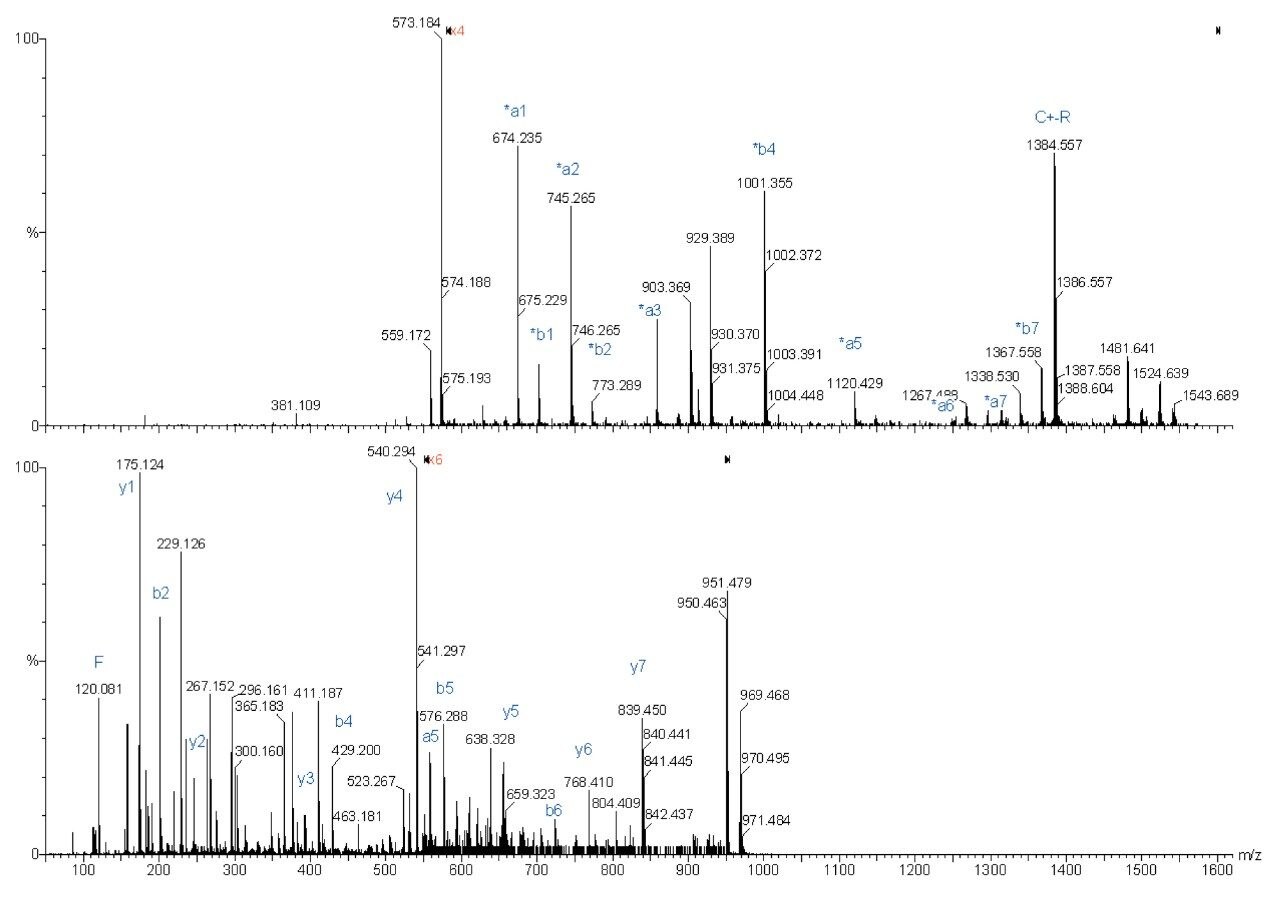
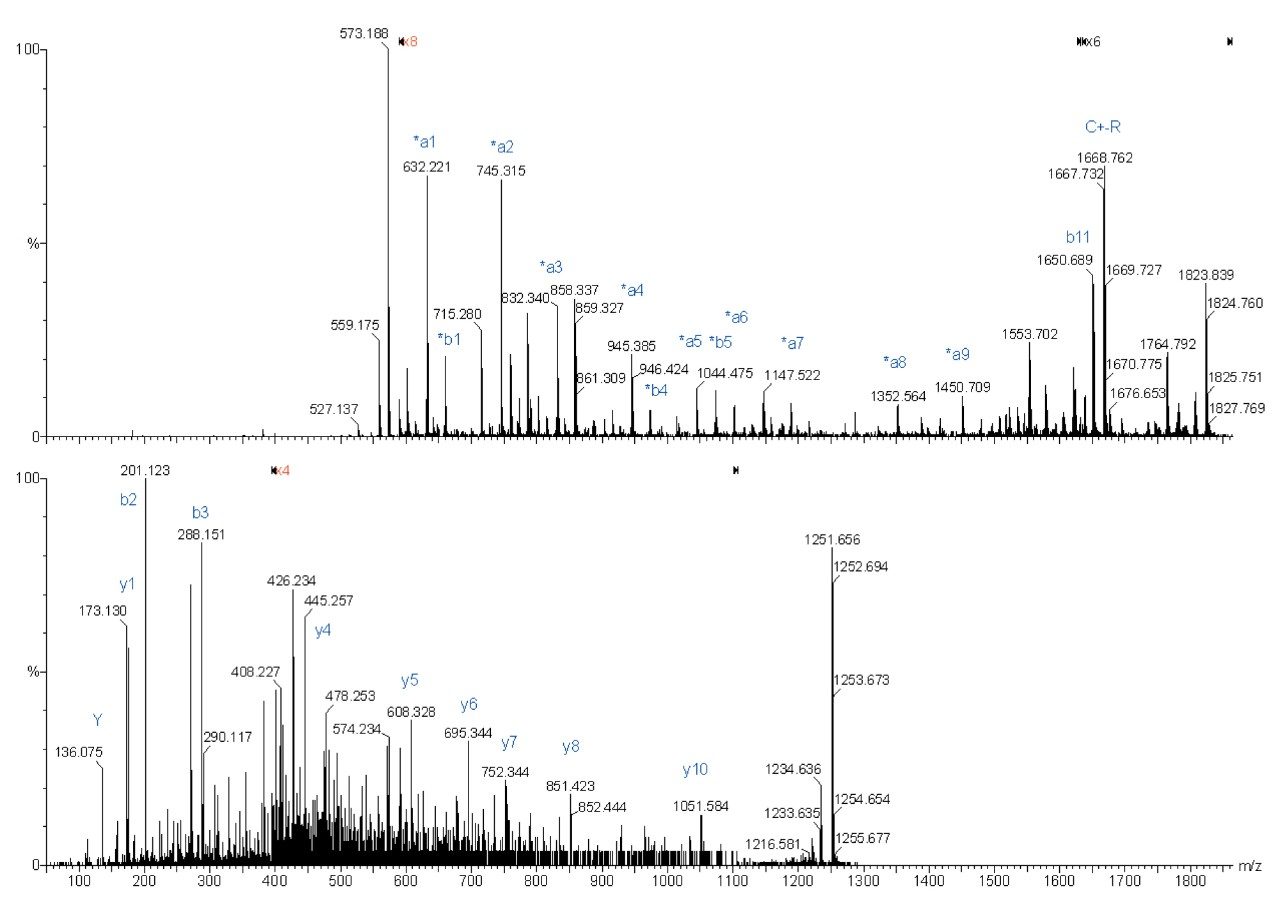
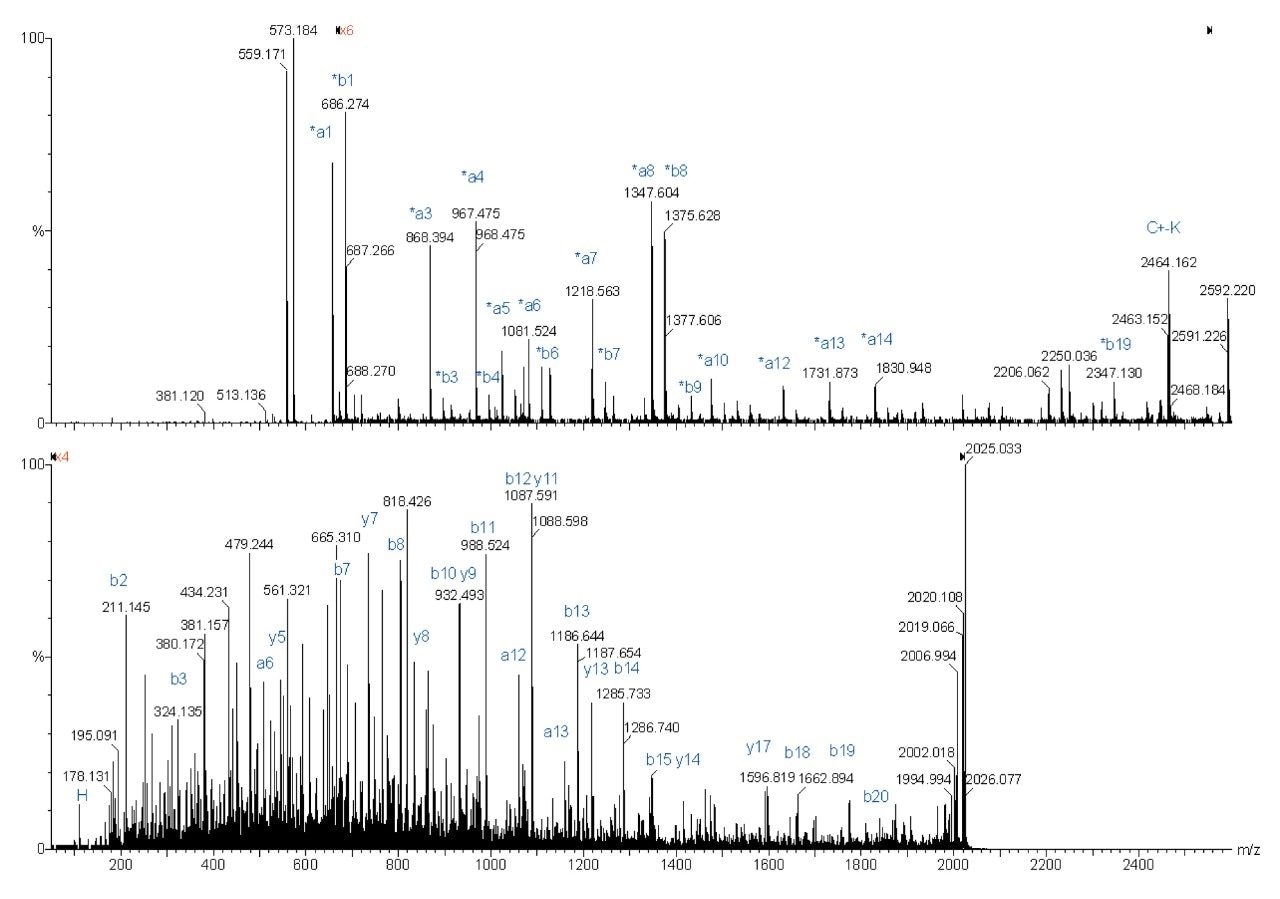
This application note describes the use of an N-terminal derivatization strategy, using the N-Tris (2,4,6-trimethoxyphenyl) phosphonium-acetic acid N-hydroxysuccinimide ester (TMPP-acOSu) to improve MALDI MS/MS fragmentation patterns. Coupling of this peptide derivatization protocol with nanoscale HPLC separation and deposition onto a MALDI target is presented.
One advantage of this derivatization reagent is, that it increases the peptide masses by 572.182 Da. This mass increase can bring small peptides into the m/z range 800–3500, the typical range used in PMF analysis. A theoretical tryptic digestion of 100 randomly selected proteins from the Swiss-Prot databank revealed that small tryptic peptides, with fewer than eight amino acid residues, account for ca 20% of protein coverage.