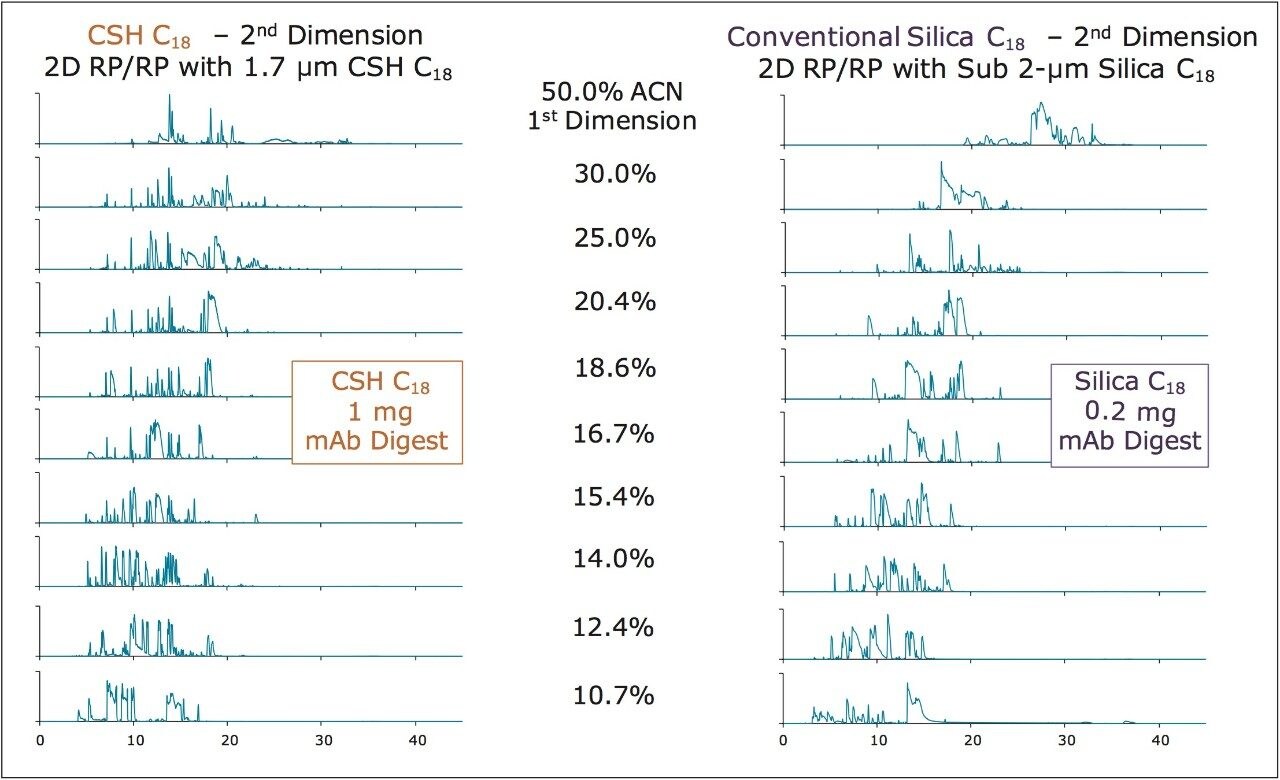
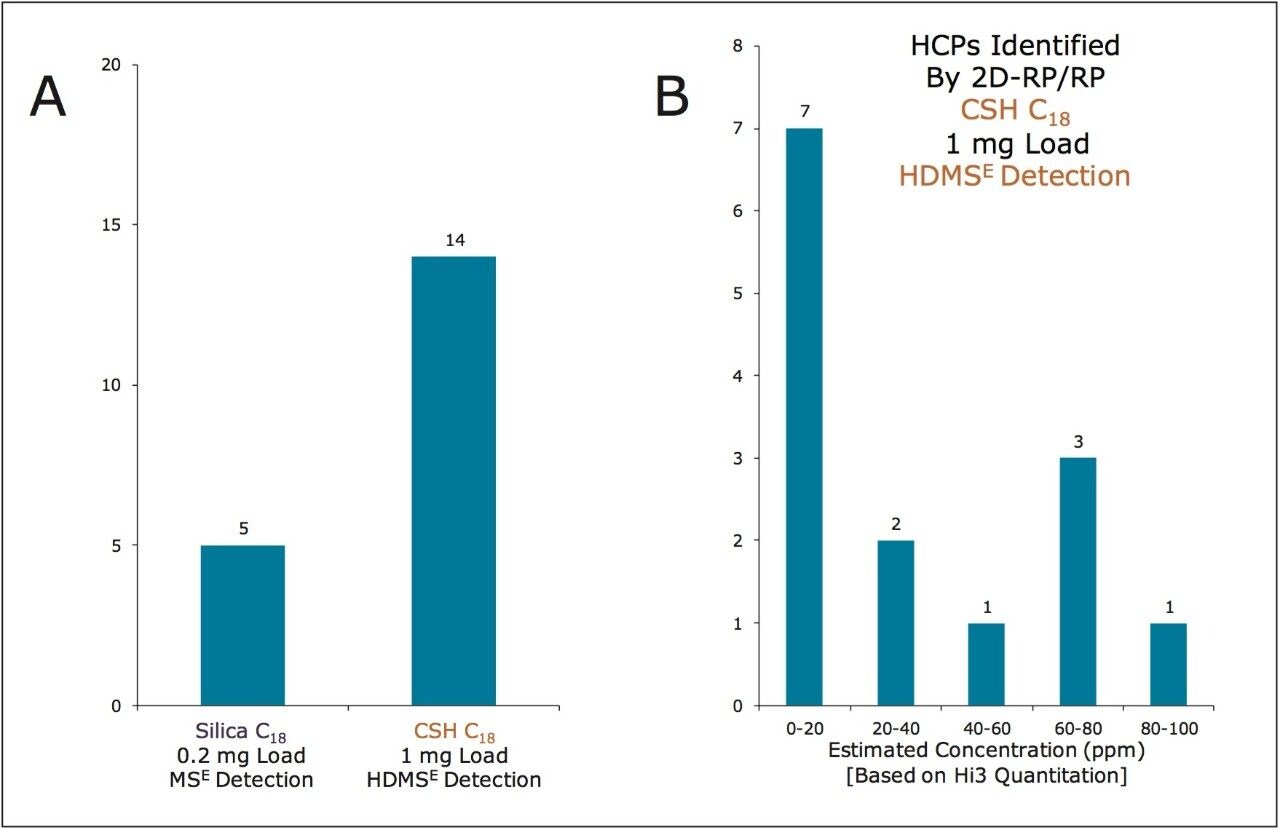
CSH C18, with its positive surface potential at an acidic pH, minimizes undesired secondary interactions, exhibits enhanced loadability, and thereby minimizes peak broadening at high mass loads.4 Such chromatographic performance is well suited to the analysis of HCPs, where there is a need to identify low-ppm impurities in the presence of highly abundant product-derived peptides that readily cause ion suppression and spectral crowding if co-elution occurs. It has previously been demonstrated that two-dimensional (high pH/low pH) RP/RP separations can be used to obtain high peak capacity peptide separations and to identify HCP impurities.1,5 Indeed, such chromatography operated at near 10K psi with an ACQUITY UPLC M-Class System and 300 μm I.D. sub-2-μm, silica-based C18 analytical column produces a theoretical peak capacity (half-height) of 2535 when a test mixture is analyzed at low mass loads.6 In practice, however, this peak capacity is substantially compromised when analyzing the mass loads required to characterize HCPs. Interestingly, the use of CSH C18 in the 2nd dimension of such a 2D-RP/RP system has been found to minimize the peak broadening observed during HCP analyses. CSH C18 thus enables even higher mass loads to be analyzed without significant losses in peak capacity, which assists in detection of impurities at increasingly lower abundances.
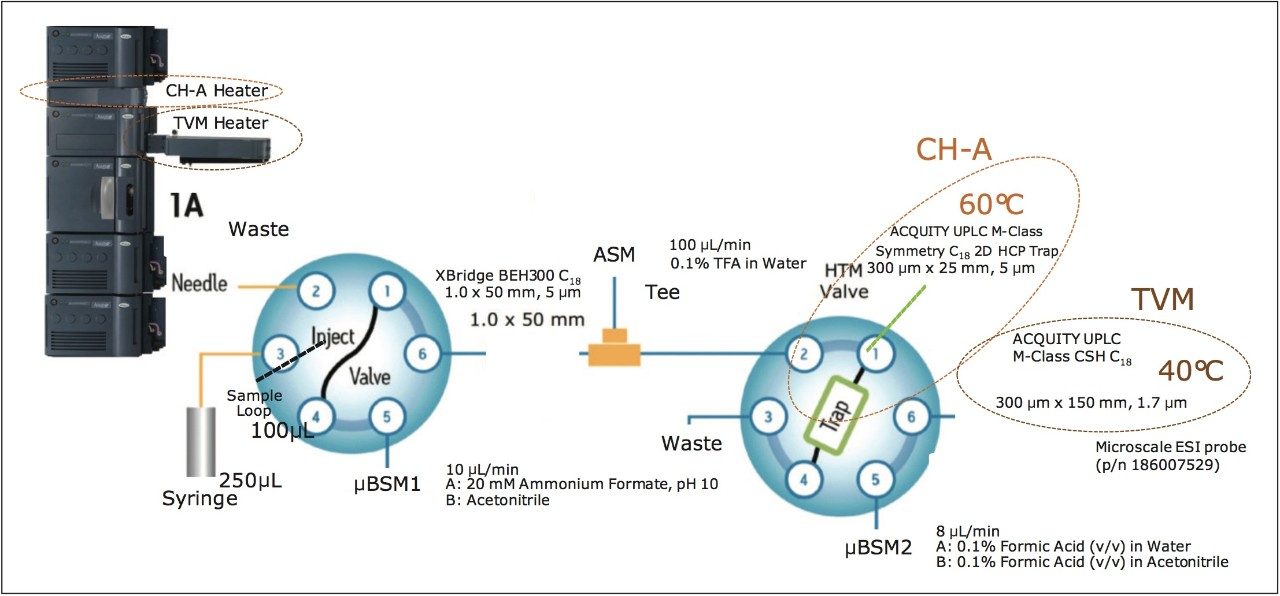
For this reason, the combination of UPLC 2D-RP/RP with a 300 μm I.D. CSH C18 column is presented for improved HCP analysis. Figure 1 shows a schematic of this apparatus, in which an ACQUITY UPLC M-Class System is outfitted with an ACQUITY UPLC CH-A Column Heater to allow for differential heating of trapping and analytical columns. Dual zone heating can be essential to optimizing trap-elute separations, such as the second dimension segment of this 2D-RP/RP system. Here, a Symmetry C18 trapping column is maintained at 60 °C to facilitate optimal refocusing on the CSH C18 2nd dimension analytical column (see Reference 7 for more information on differential column heating).7