To demonstrate the advantages of appropriate sample preparation, the therapeutic monoclonal antibody trastuzumab was analyzed in human plasma. Two parallel strategies were used. In one scheme, 96-well protein A purification plates were used to isolate the IgG fraction (including trastuzumab) from plasma samples. In a second scheme, no protein level cleanup was performed. Each set of samples was then denatured with RapiGest, reduced, alkylated and digested with trypsin to yield peptide fragments suitable for LC-MS/MS analysis. These resulting digests were then split into two aliquots. One aliquot was directly analyzed by LC-MS/MS while a second was further cleaned up using Oasis MCX µElution plates prior to LC-MS/MS analysis.
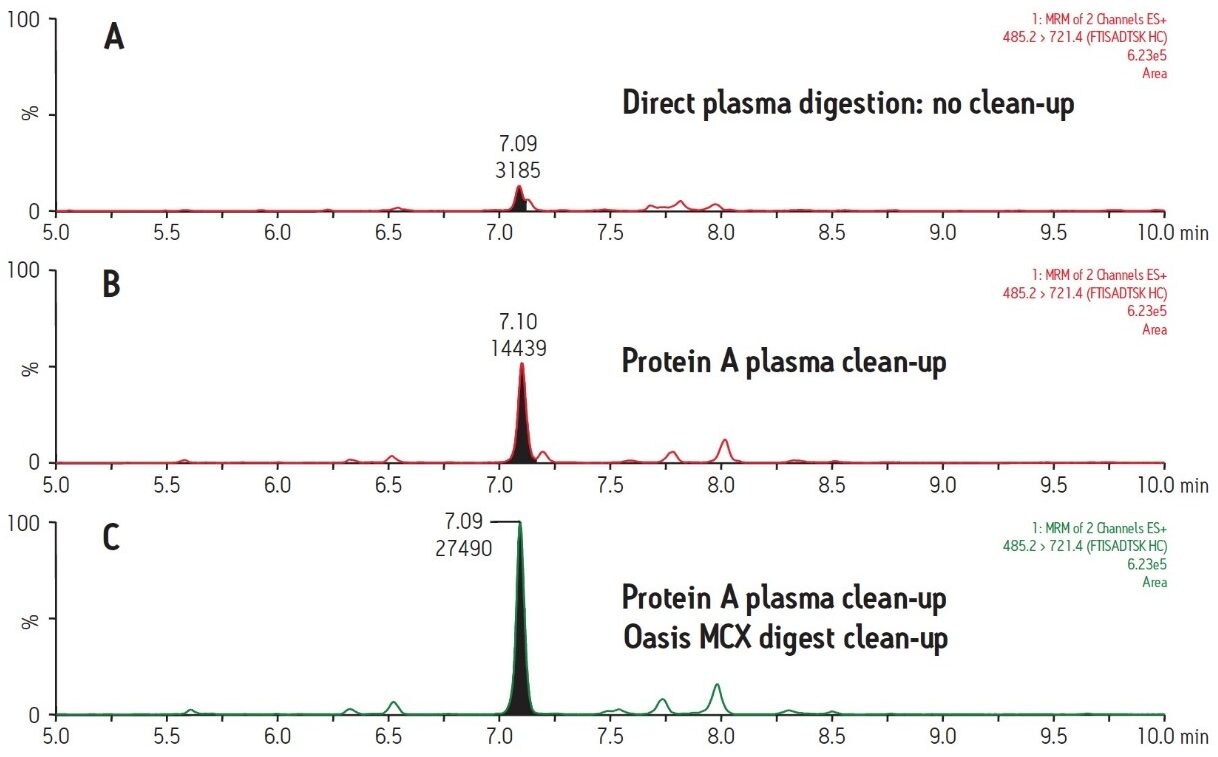
Figure 2 demonstrates the differences and incremental benefits achieved through various degrees of sample preparation. The sample in Figure 2A was a direct digestion of the plasma, followed by direct injection of the subsequent digest. There was no sample cleanup performed at either the protein or peptide level. The resultant chromatogram yields a poor intensity peak which co-elutes with closely related interferences, rendering accurate quantification challenging and yielding a poor LLOQ. The sample in Figure 2B was cleaned up at the protein level only using protein A 96-well plates. This type of sample prep is generic enough to be applied in any workflow where the drug is an IgG-based antibody, making it ideal for a discovery setting. Furthermore, the 96-well format is also compatible with high-throughput analysis. The increase in peak area for the signature peptide relative to no protein-level isolation (Figure 2A) is nearly 5 fold. The chromatogram in Figure 2C represents a sample which has been cleaned up both at the protein level with protein A and at the peptide level with mixed-mode cation exchange SPE. The incremental benefit over protein A cleanup alone (Figure 2B) is additional 2 fold increase in peak area and an elimination of adjacent peaks, increasing specificity of the assay. Mixed-mode SPE cleanup not only removes interfering peptides, but also removes digest reagents, buffers, and other plasma components such as phospholipids, improving instrument robustness and data quality. Protein A sample preparation also eliminates high abundance plasma proteins such as albumin and transferrin, which reduces the required SPE capacity and facilitates the use of the µElution plate format. The major benefit of this SPE format is concentration of the sample without evaporation, thus minimizing peptide losses due to adsorption. Although not tested here, mixed-mode SPE can also be used to purify signature peptides from direct plasma digests (without protein-level cleanup). However, a larger bed mass 96-well plate may be required to accommodate the same sample volume due to the increased concentration of undesired peptides. An additional advantage to the use of generic protein level isolation, such as protein A, is that it reduces the required amount of trypsin by almost 10X, significantly reducing the assay cost.