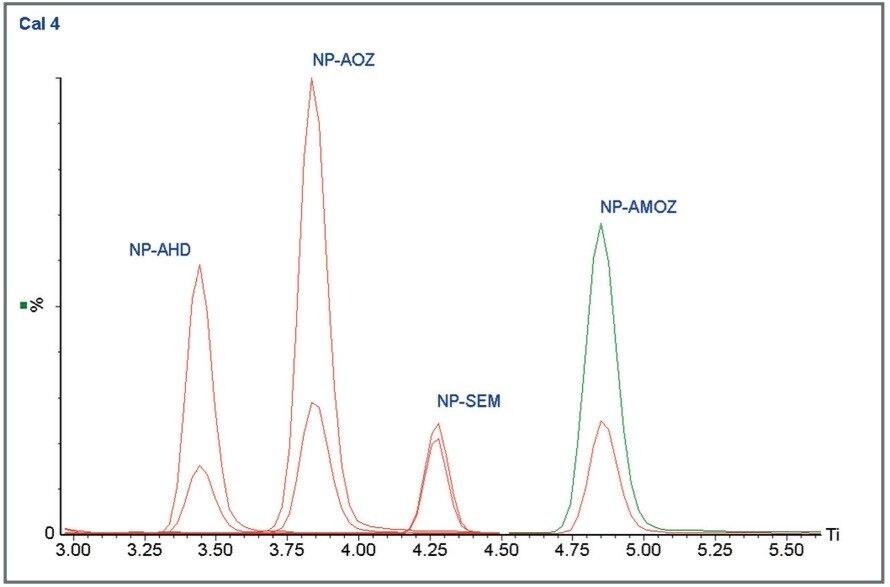
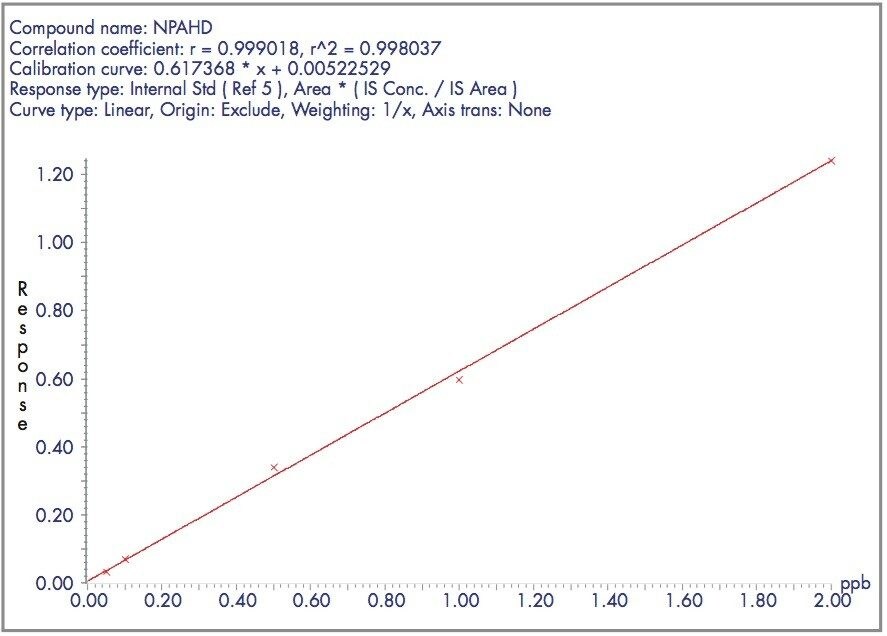
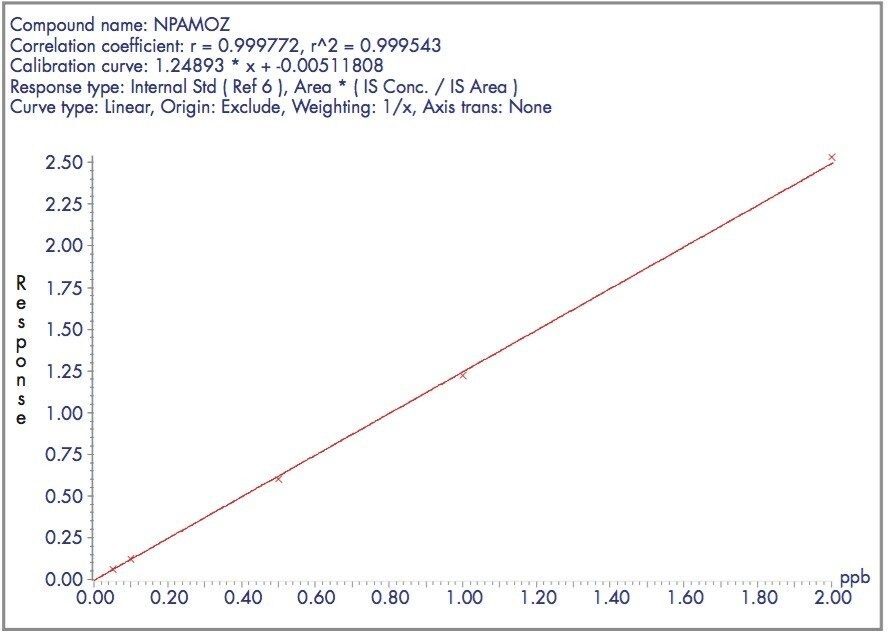
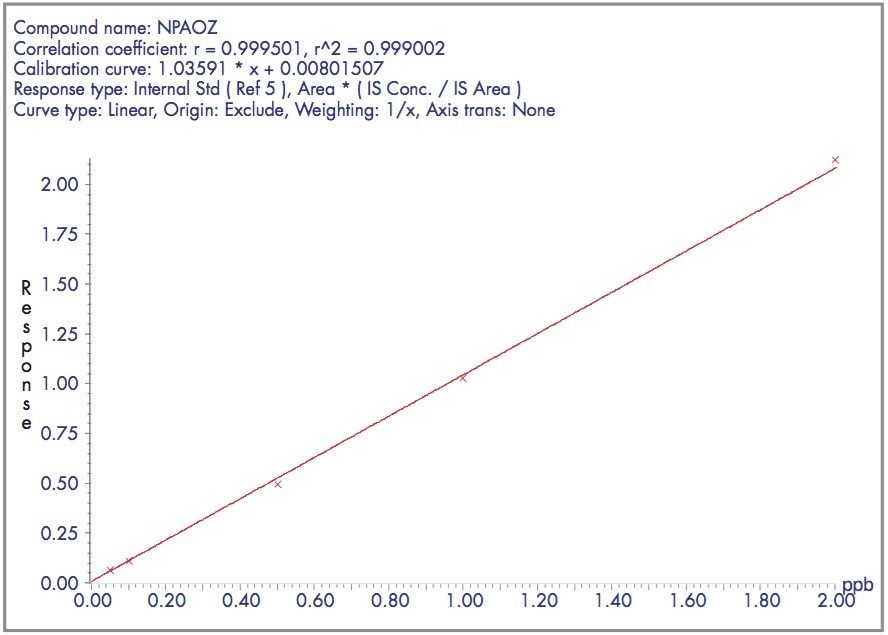
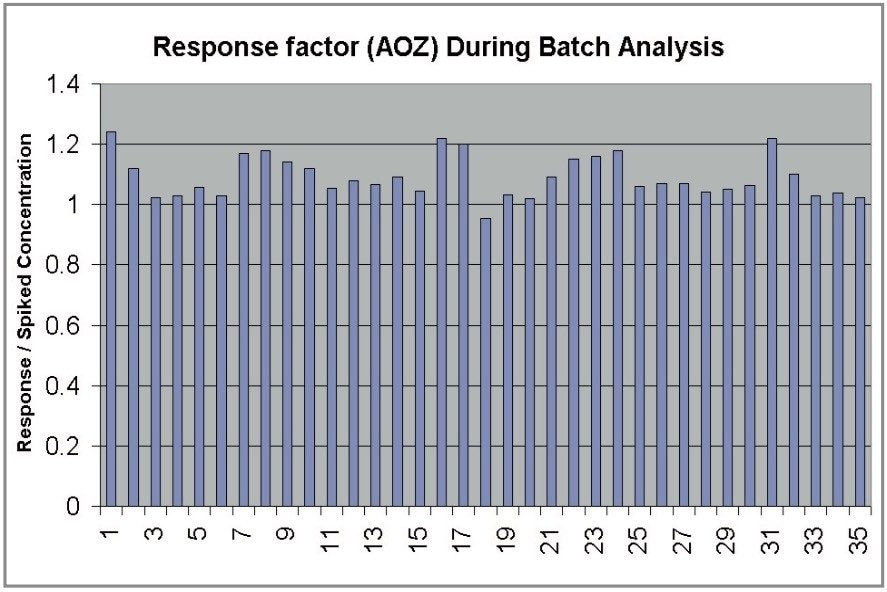
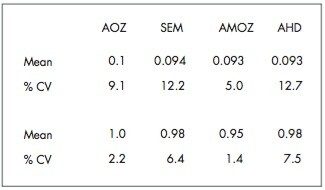
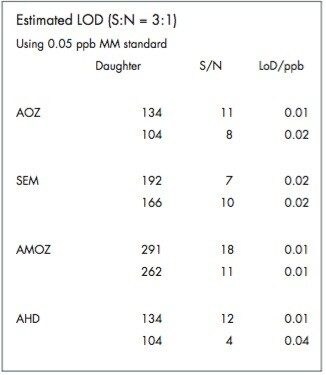
The four nitrofuran antibiotics furazolidone, furaltadone, nitrofurazone and nitrofurantoin are banned from use in the medication of animals destined for human consumption.1 Once administered to livestock, the drugs are rapidly metabolized and, after a few hours, cannot be detected in edible tissues. Certain metabolites of these drugs are more persistent and their presence can be used as a marker of illegal use. Proportions of these metabolites exist as protein adducts and the established method of analysis contains an acid hydrolysis step prior to extraction. A less complex sample matrix may be obtained if the homogenized sample tissue is washed with organic solvents prior to the hydrolysis step. Such a method is used to target only the bound residues since any free metabolite will be removed during the washing stages.2 This has the advantage of reducing sample matrix effects and minimizing the amount of routine instrument maintenance required.
Previous Waters application notes have described the analysis of nitrofuran metabolites using the Waters Micromass Quattro Ultima Platinum Mass Spectrometer.3 The Quattro Premier is a tandem quadrupole mass spectrometer that incorporates novel travelling wave (T-Wave) ion transfer and collision cell optics, together with improved detector technology in order to deliver unsurpassed sensitivity and method flexibility.4 This note describes the performance of the Quattro Premier Mass Spectrometer for the analysis of bound nitrofuran metabolites in chicken meat.