The results demonstrate that UPLC-MSE is a suitable tool for characterizing PTMs in monoclonal antibodies. MSE ensures sampling of low-abundance components and acquires indiscriminately MSE spectra, enabling accurate identification of modified peptides in an unbiased, reproducible manner. The specific conclusions from this study:
1. UPLC-MSE is capable of separating, identifying, and quantifying modified peptides and isoforms
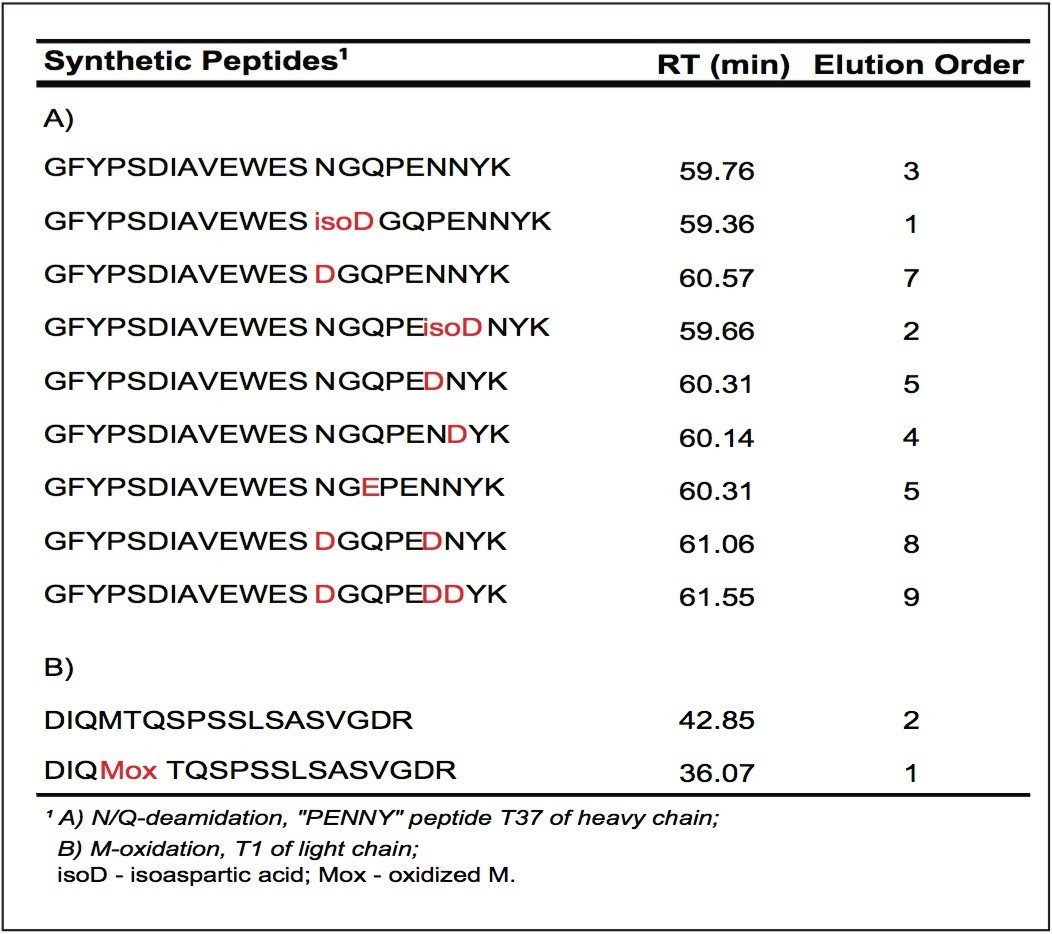
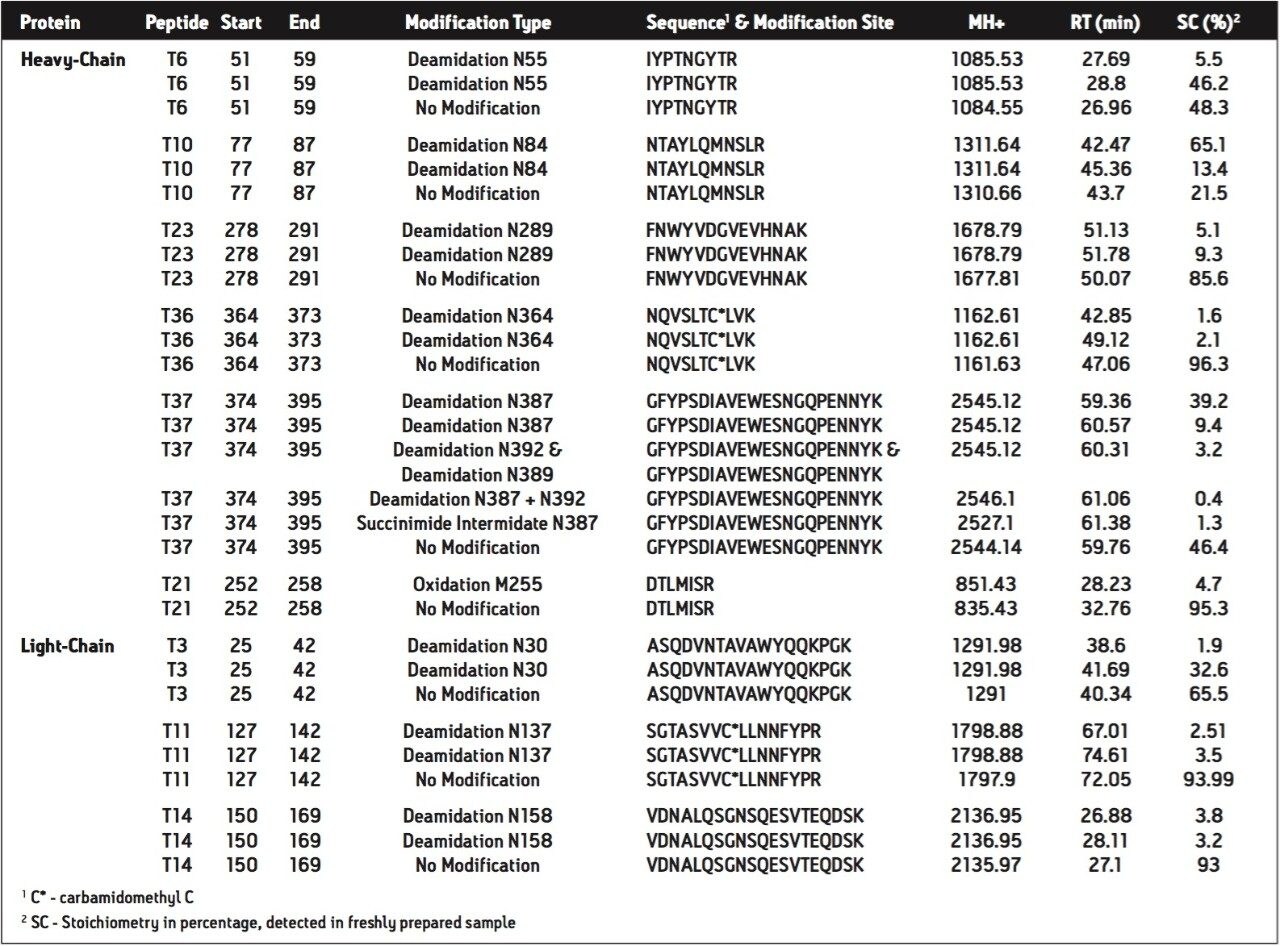
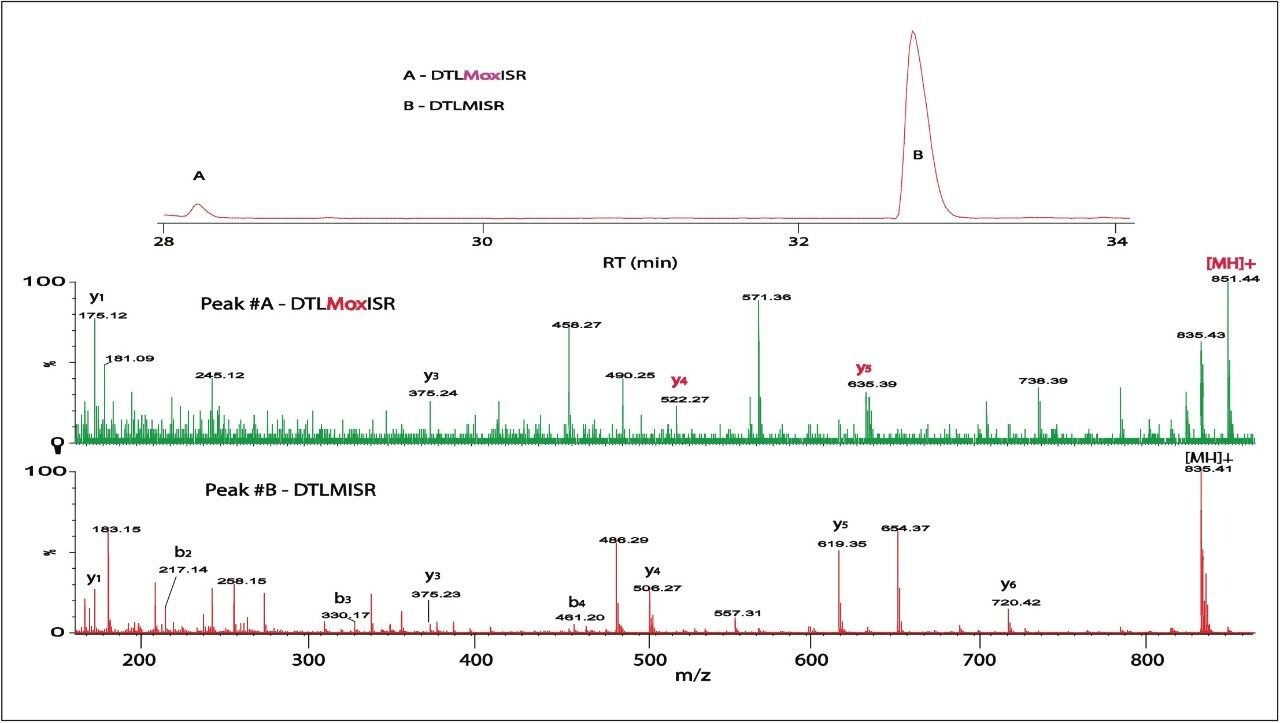
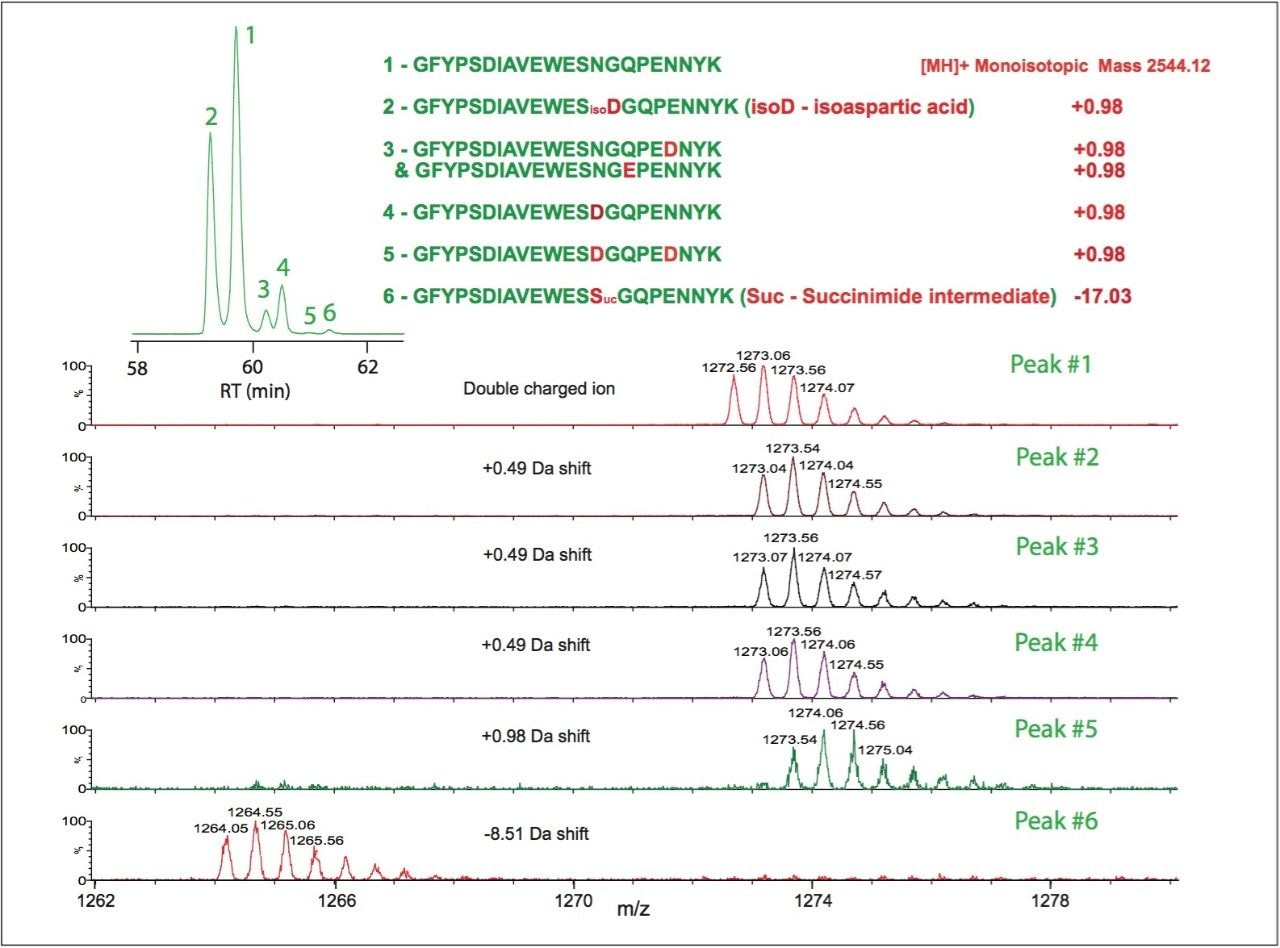
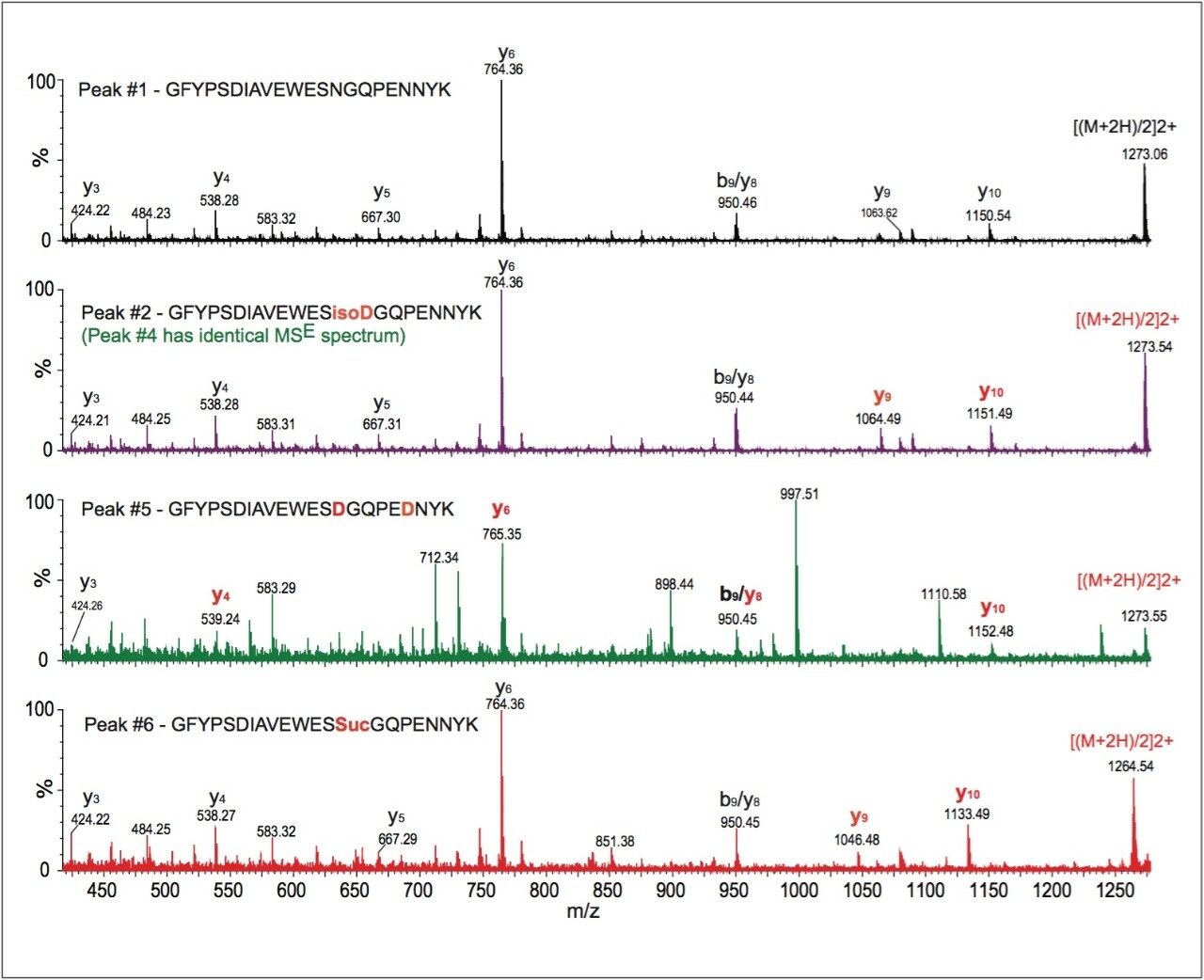
2. The high mass resolution and high mass accuracy of the SYNAPT MS System ensures confident identification of modifications with small mass shift (e.g., N-deamidation with 0.98 Da mass difference) and modified isoforms
3. Synthetic peptides are helpful for determining modified isoforms and are required for confirmation
In a previous study4, we have demonstrated that UPLC-MSE is able to provide high sequence coverage mapping of mAb tryptic digest, with 97% sequence coverage for both light and heavy chains of the antibody. Therefore, UPLC-MSE and SYNAPT MS system is an advanced platform for characterization of recombinant proteins, such as monoclonal antibodies.
In current LC-UV/MS peptide mapping methods, the identification of peptide sequences and determination of site-specific modifications typically require multiple tandem mass spectrometry experiments (either DDA MS/MS or targeted MS/MS). The methodology reported here achieves both goals in a single LC run because of available peptide fragmentation information provided by MSE.
Peptide mapping with UPLC-MSE improves the analytical efficiency of peptide characterization.