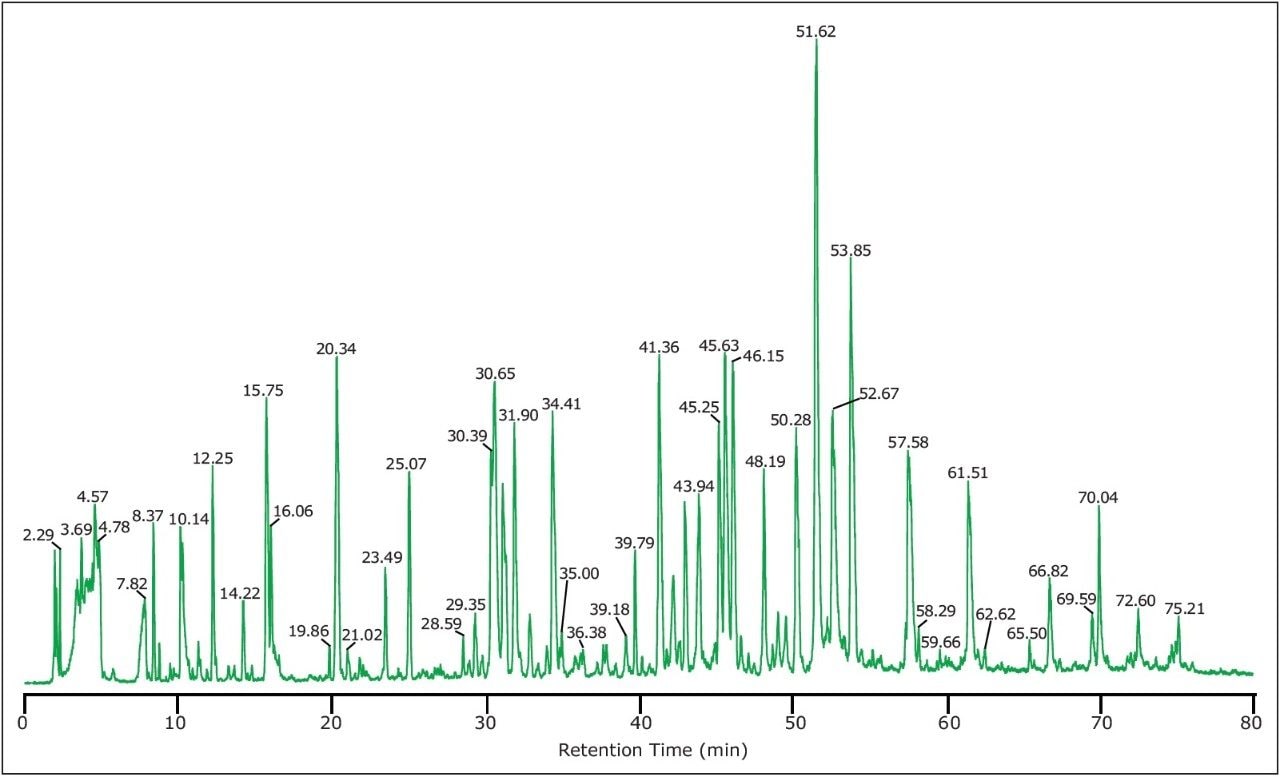
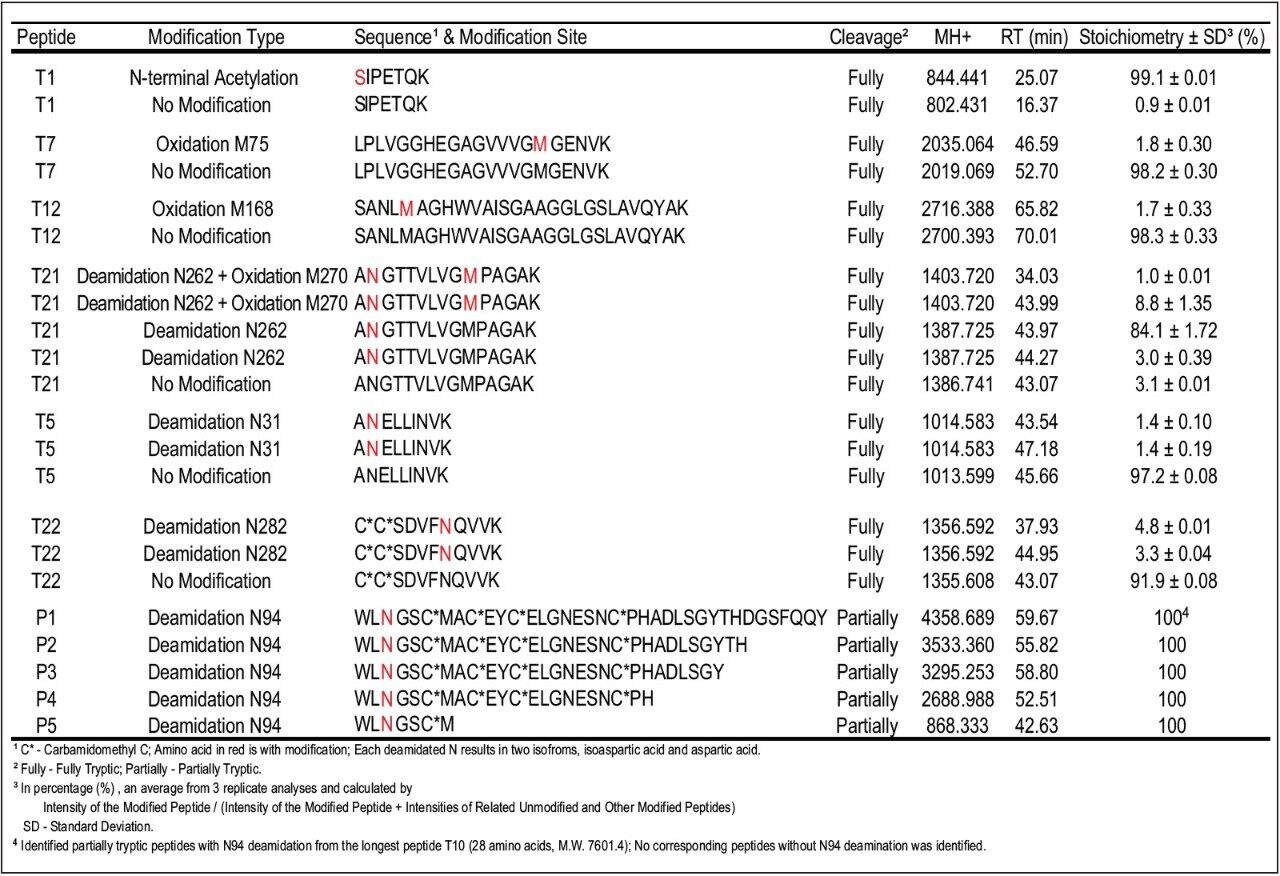
The relative quantification of modified peptides calculated from MS signal intensity (Table 1) shows that over 99% of N-terminal of ADH1 were found to be acetylated. Three methionines in the protein sequence were found to be oxidized to a relatively low degree, less than 2% for M75 and M168 sites, and approximately 10% for M270 site. In addition to M-oxidation, the peptide T21 was nearly completely deamidated at N262 site (97%), present in four isoforms as shown in Table 1. Similarly, high percentage of deamidation was observed for N94 site.
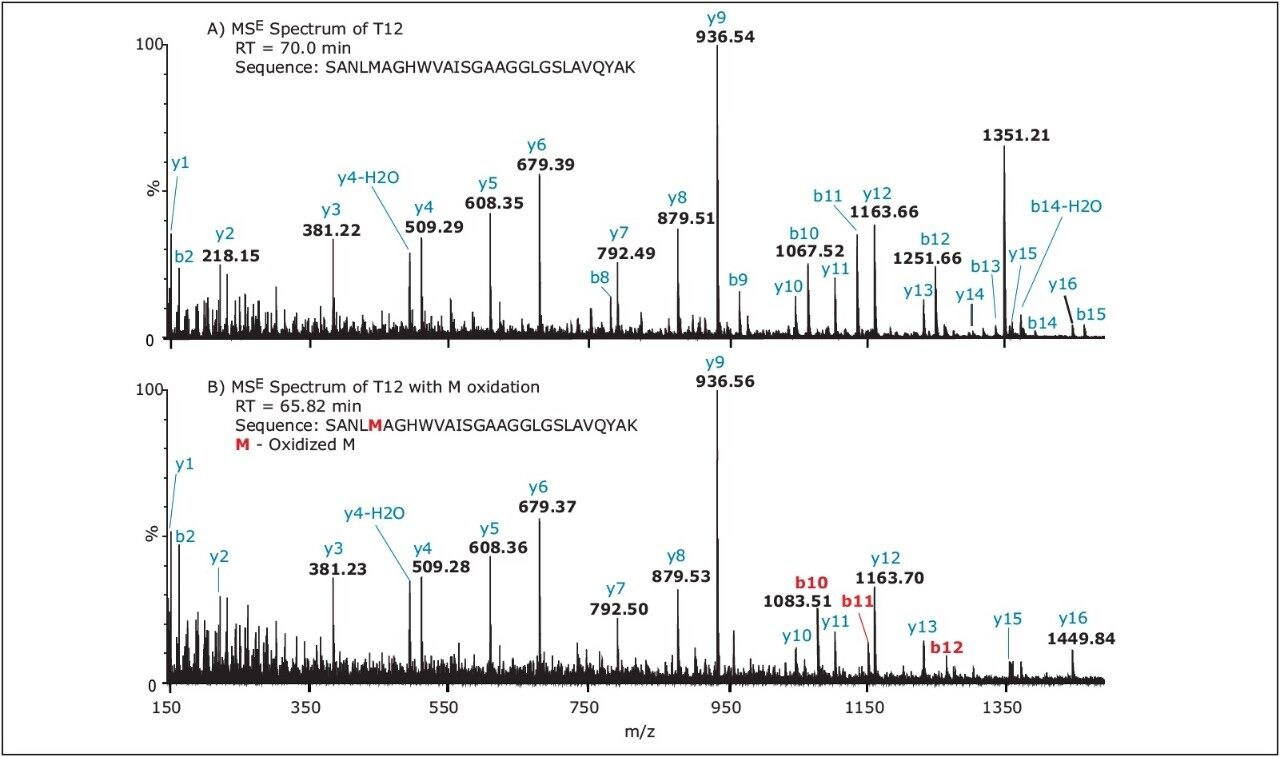
Although the tryptic peptide T10 (consisting of 69 amino acids, molecular weight 7601.4) was not detected, a series of partiallytryptic N94-deamidated peptides covering part of the missing T10 sequence were found (P1, P2, P3, P4, P5 as shown in Table 1). The non-deamidated versions of these peptides were not detected. Finally, the deamidation of N31 and N282 sites was found to be less than 10%. Peptide RT shift upon modification can be used as an additional confirmation of sequence modification. In general, N-terminal acetylation increases, while M-oxidation decreases the peptide retention. The RT of N-terminal peptide T1 shifted from 16.37 min to 25.07 min after N-terminal acetylation. In contrast, the RT shifted from 52.7 min to 46.59 min for peptide T7 and from 70.0 min to 65.82 min for peptide T12 after M-oxidation in these peptides.
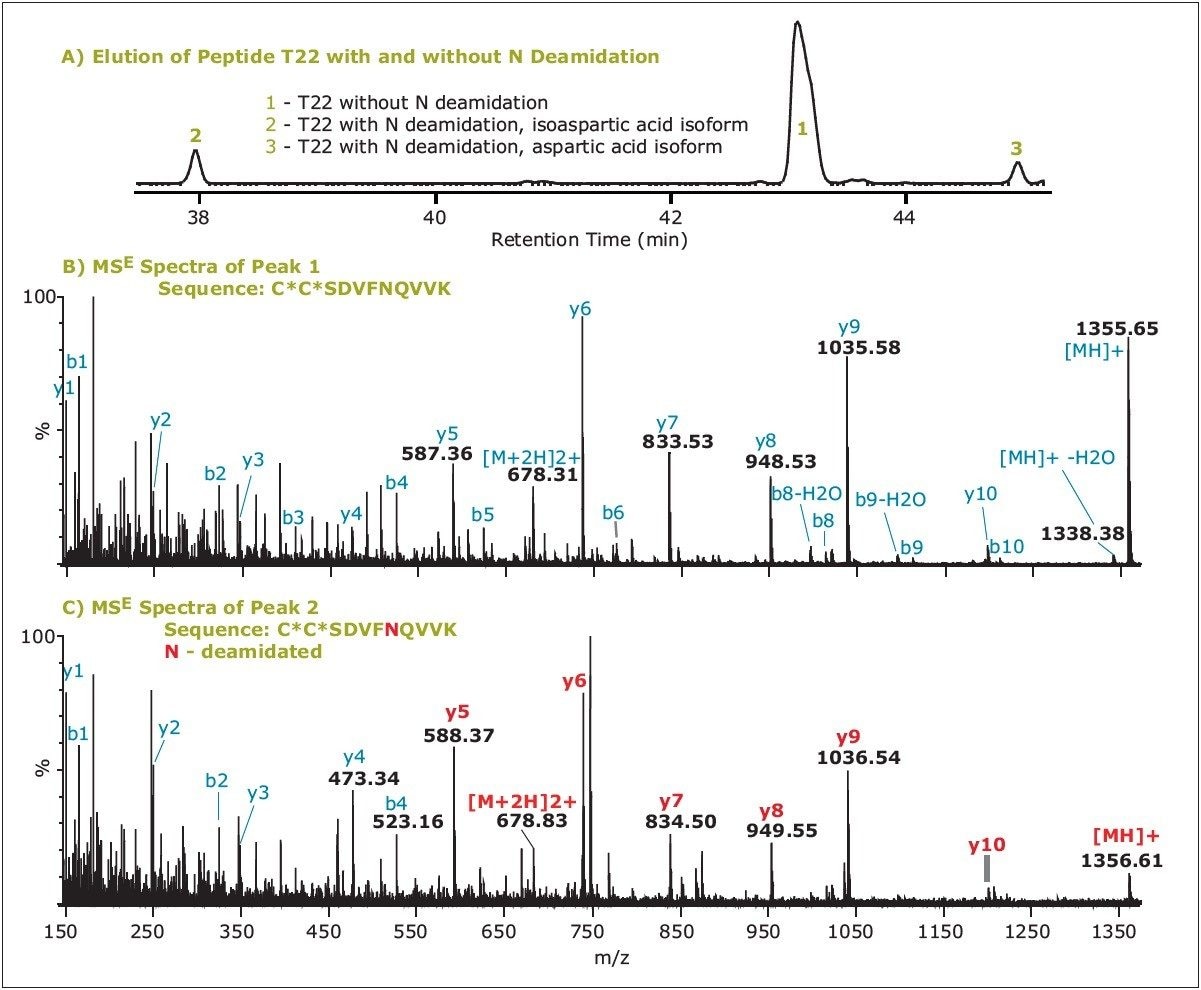
In the case of N-deamidation, the RT trends are more complex because of the presence of two product isoforms: isoaspartic acid and aspartic acid. Usually, N-deamidated peptide with isoaspartic acid elutes earlier and the other deamidated one with aspartic acid later than the unmodified peptide. There are exceptions to this rule, but the retention order observed here was always the isoform with isoaspartic acid < aspartic acid isoform.
The retention pattern of unmodified peptide T22 and its two N282-deamidated isoforms is presented in Figure 3A. Since the two deamidated isoforms are isobaric (both +0.98 Da mass difference from the unmodified T22), they cannot be distinguished from MS or MSE data. In such cases, the UPLC separation and RT information of these peptides are important for the identification. However, MSE can easily differentiate N-deamidated peptide from unmodified peptide, as shown in Figure 3, with a high mass resolution and high mass accuracy platform such as the SYNAPT MS System.