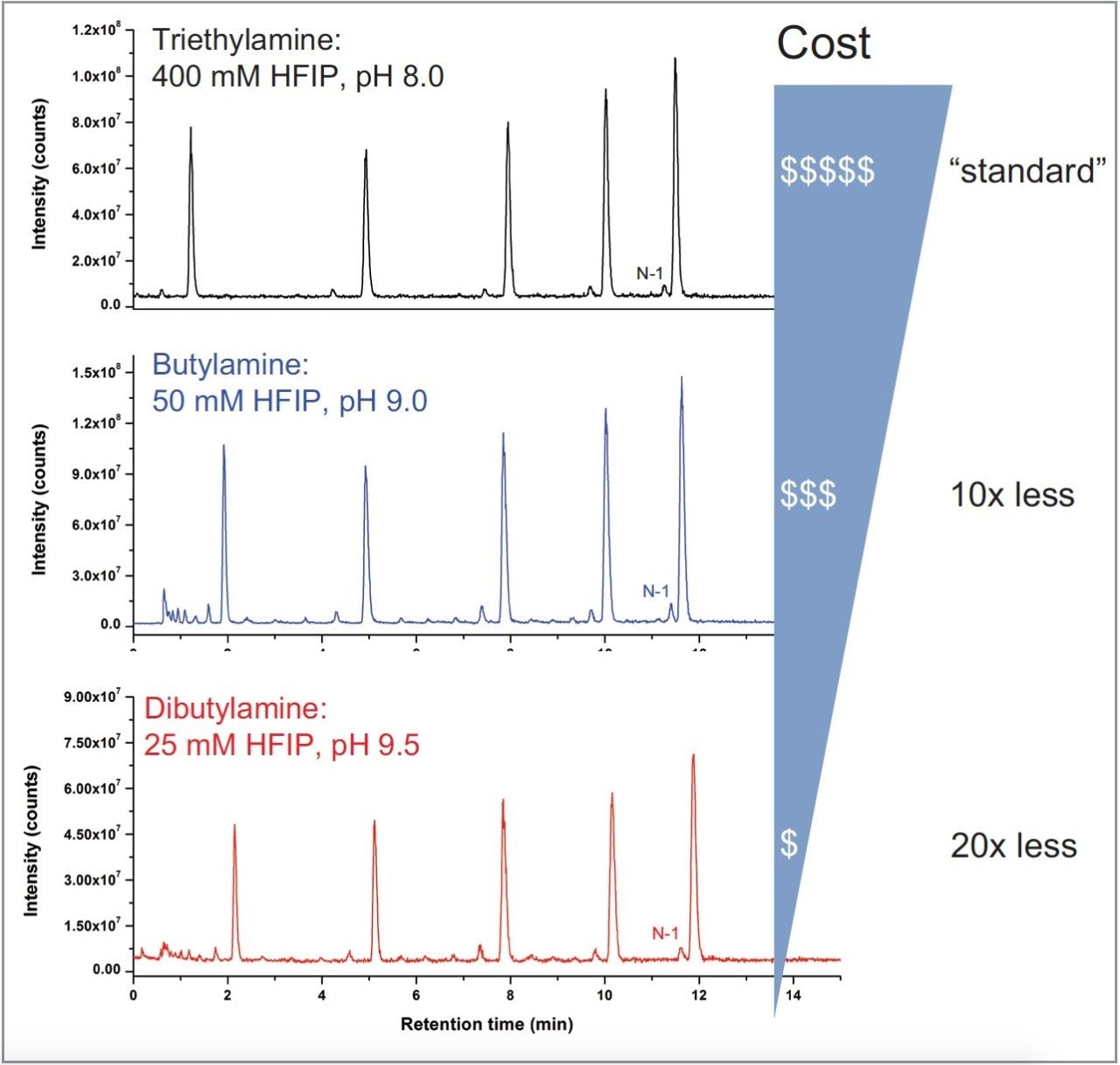
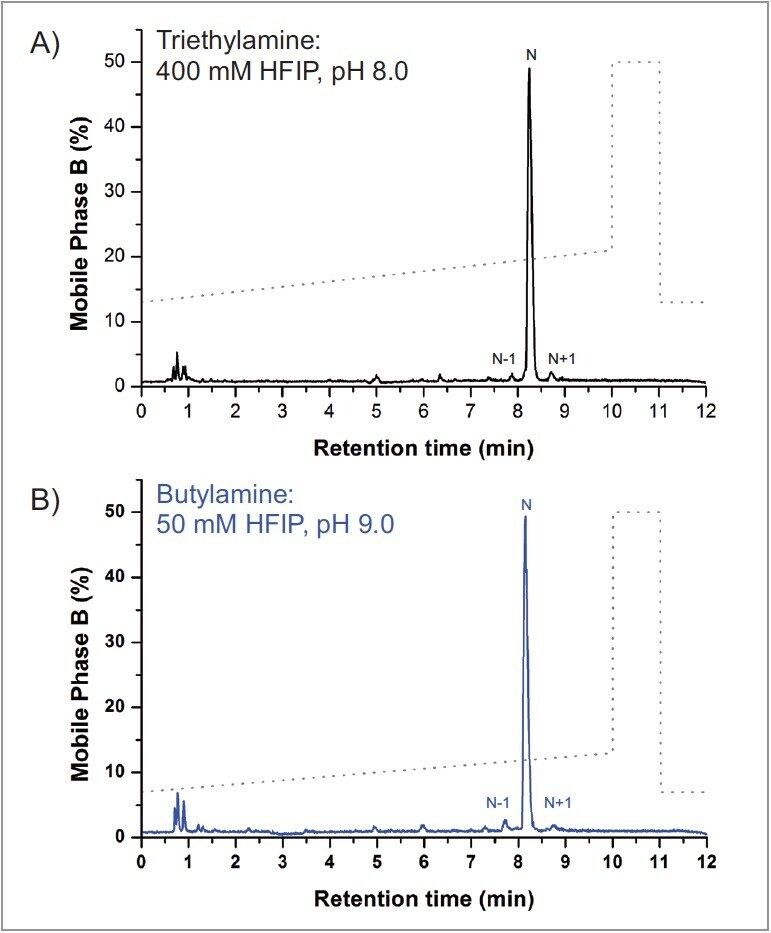
As shown in Figure 1, comparable selectivity was achieved with the two alternative amines. Furthermore, butylamine was able to achieve similar selectivity as the TEA:HFIP with a gradient of 0.46% B/min, but with a lower starting organic composition; whereas dibutylamine required doubling the gradient to 0.80% B/min with similar starting organic composition as TEA:HFIP. These results suggest the concentration of HFIP and hydrophobicity of the amine impact oligonucleotide retention on the column. However, “tuning” of the separation conditions can be performed to achieve the desired separation, which in this case was to match the selectivity of the TEA:HFIP separation.
Mass detector response was shown to be similar between TEA:HFIP and BA:HFIP, with BA:HFIP showing marginally better signal response. This was in contrast to the DBA:HFIP run, which showed approximately a 2-fold drop in signal intensity, despite having an identical mass load. The reduced signal intensity in the DBA:HFIP separation does not hinder detection of the failed sequence peak (N-1, Figure 1), indicating alternative amines such as butylamine and dibutylamine can produce adequate MS signal response for analysis when buffered with HFIP.
Interestingly, an unintended corollary with assay cost was observed during the comparability test. Cost-prohibitive MS-grade purity reagents such as HFIP are often cited as a concern in oligonucleotide assay development. Figure 1 demonstrates that IP reagents such as butylamine and dibutylamine can reduce assay costs through reduced HFIP use while maintaining assay selectivity when compared to TEA:HFIP, making them an appealing alternative in IP-RPLC/MS-based techniques. As indicated in Figure 1 though, lowering the concentration of HFIP results in a mobile phase with a pH >8.0, which can be a concern with respect to column longevity.
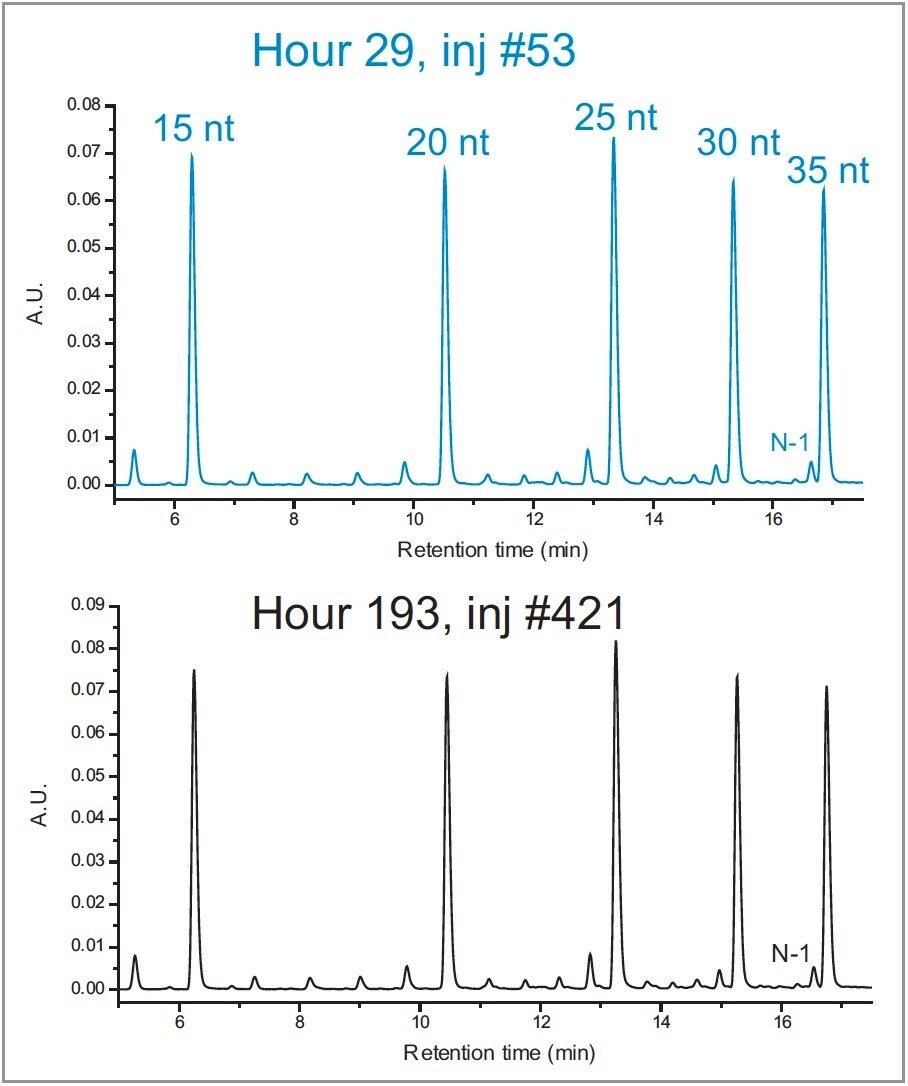
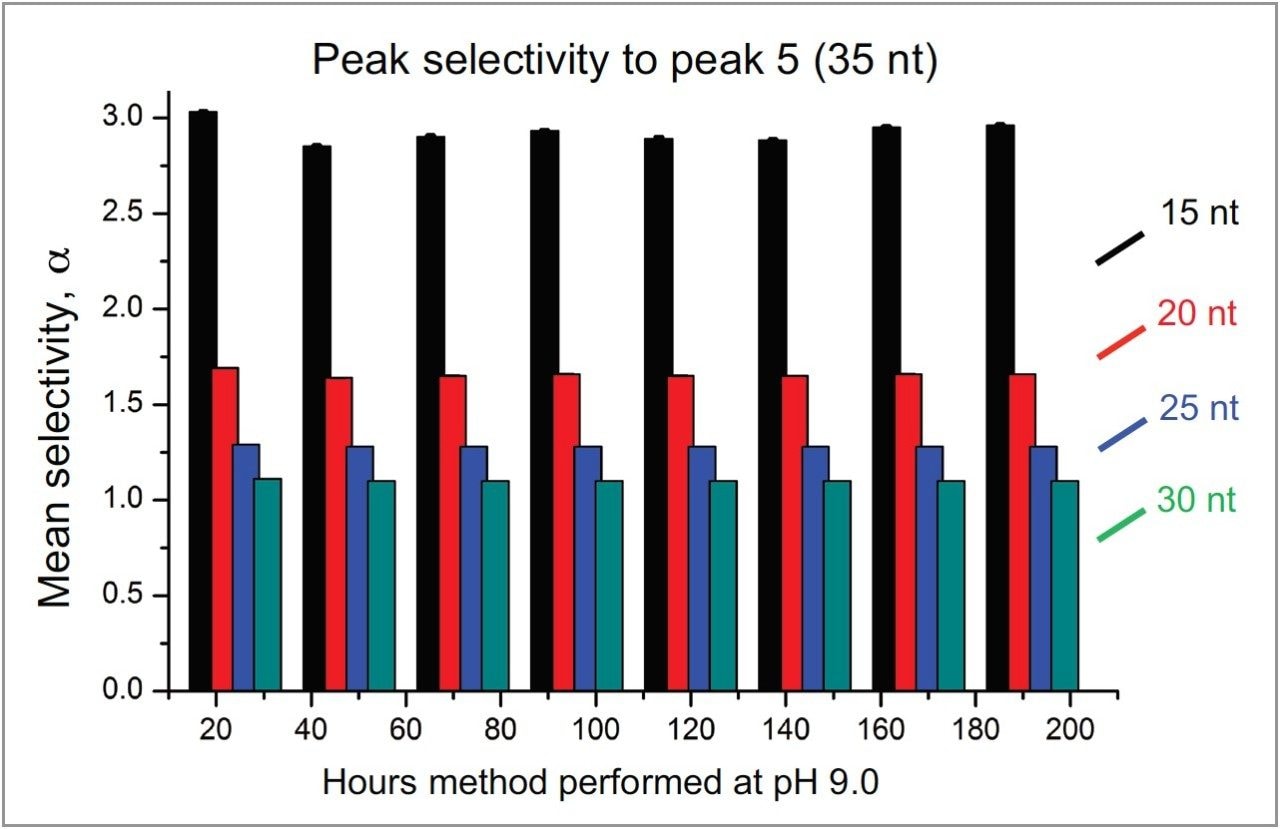
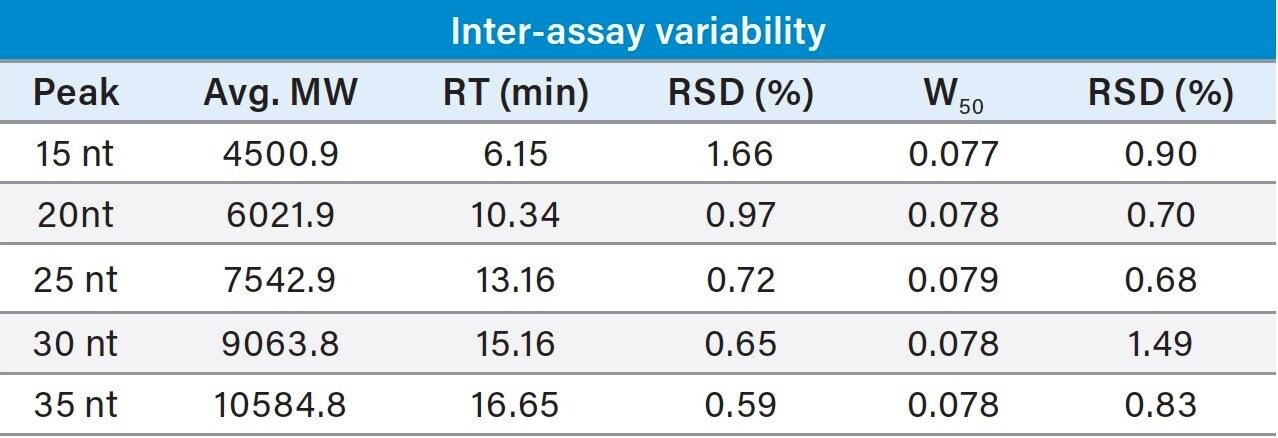
To test the impact of a modest increase in pH on column longevity, the mobile phase prepared with butylamine (15 mM BA:50 mM HFIP, pH 9.0) was selected for evaluation. For this study the gradient was extended to 30 minutes with a slope of 0.5% B/min. Mobile phases were prepared in 200 mL batches and refreshed every 24 hours over the course of the time study. To conserve samples, four water blanks were injected in succession using the method followed by an injection of the mixture of polyT standards. As shown in Figure 2, the PolyT standards were separated with a high degree of repeatability over 400 injections with nearly identical chromatographic profiles. Selectivity was calculated for the first four peaks relative to the last peak as a means to probe column robustness in the presence of elevated pH and temperature. Calculated values based on the experimental data were averaged over 24-hour time blocks and reported with corresponding error bars as shown in Figure 3. Selectivity values were determined to have less than 1% RSD across 400 injections over 200 hours.