Quantitative results
Calibration and quality control samples were prepared as previously described in the materials and methods section. Calibration ranges were from 0.1–100 ng/mL for THC-OH and THC-COOH, and 0.05–100 ng/mL for THC. Quality control samples were prepared at low, medium, and high concentrations as appropriate for the calibration ranges.
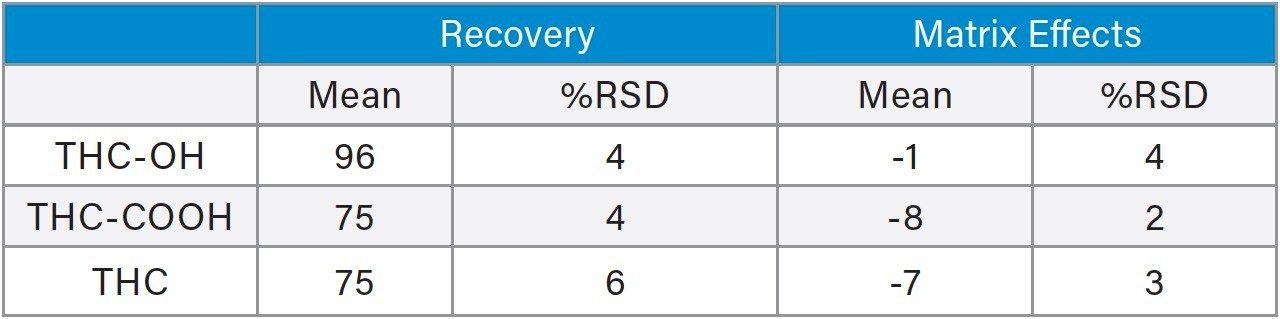
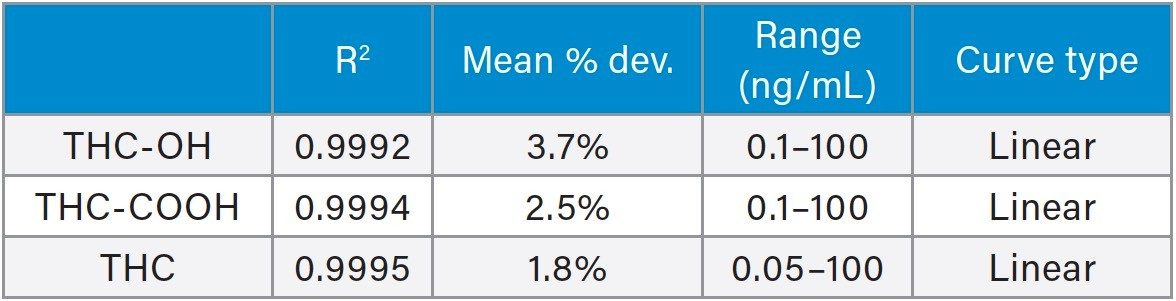
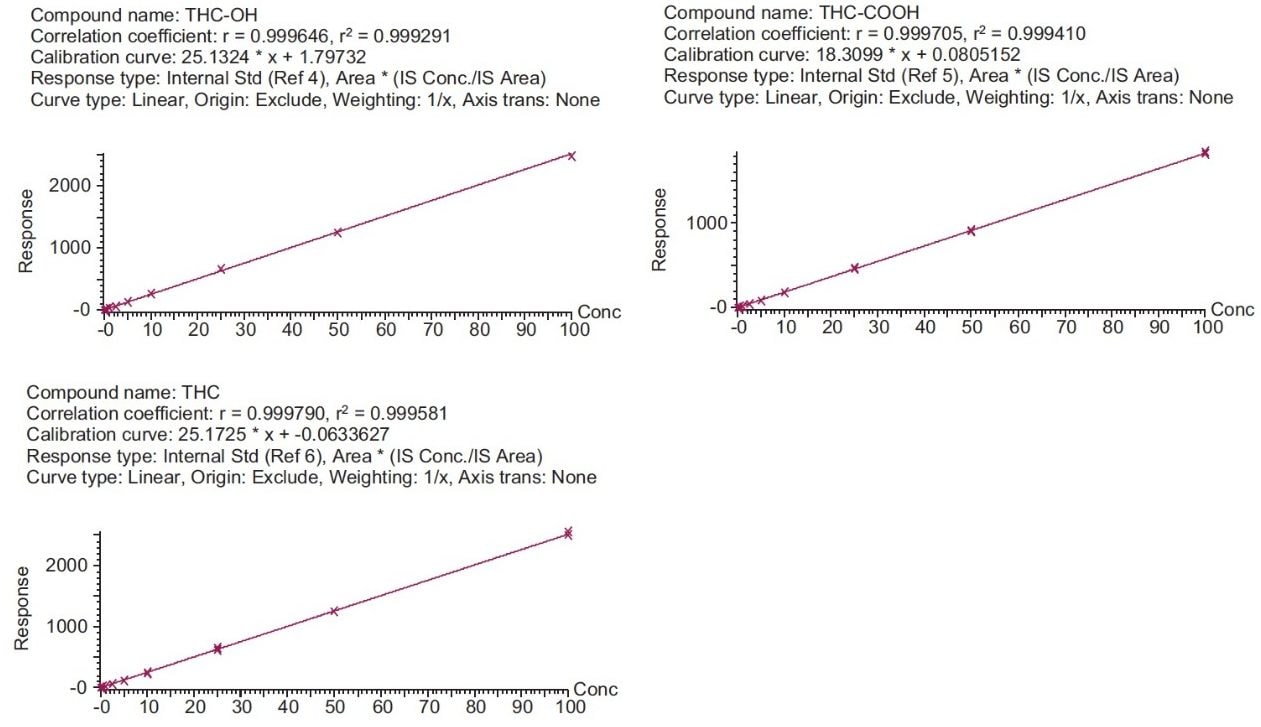
Calibration and quality control (QC) results indicate that this method is linear, accurate and precise. All compounds had linear responses over the entire calibration range with R2 values of 0.999 or greater using 1/x weighting. Figure 4 shows the calibration curves and Table 4 summarizes the data from these curves for all the compounds. Lower limits of quantification (LLOQ) were 0.1 ng/mL for THC-OH and THC-COOH and 0.05 ng/mL for THC. In each case, all FDA recommendations for accuracy, precision, linearity and analytical sensitivity were met for validated methods.4
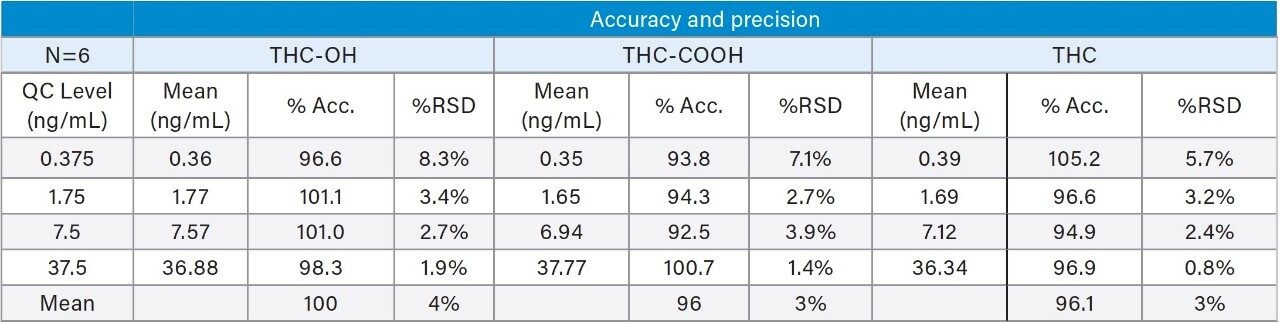
Quality control samples prepared at 0.375, 1.75, 7.5, and 37.5 ng/mL were accurate and precise. All QC values were within 10% of their target values, and most were within 5%. This data can be seen in Table 5. This demonstrates that the method is linear, accurate and precise over a calibration range that includes the entire scope of expected values of samples. The method was also proved to be both selective and sensitive enough to routinely measure THC in oral fluid well below 2 ng/mL cut off level. This was exemplified by the excellent accuracy and precision at the 0.375 ng/mL QC sample level, where calculated concentrations of all six replicates were within an average of 6% of expected.