The use of peptides and proteins as therapeutic agents has increased significantly in recent years. Thus, the demand for their analysis for toxicokinetic and pharmacokinetic studies is increasing as well.
Historically, biologics have been quantified using ligand binding assays (LBAs). However, with recent advances in mass spectrometry (MS) and liquid chromatography (LC) technologies current approaches towards peptide quantification in biological fluids now include LC-MS/MS. This is in part driven by the fact that LBAs can suffer from significant cross-reactivity issues and lack of standardization. Additionally, LC-MS/MS also has the advantage of greater accuracy and precision, broader dynamic ranges, specificity, and speed of method development. However, accurate quantification of peptides by LC-MS/MS is often not without its own challenges. Peptides have diverse pharmacokinetic profiles, often low circulating plasma levels (pg/mL), generally low MS sensitivity, and require chromatographic resolution from endogenous isobaric matrix interferences.1 Therefore, to achieve low pg/mL quantification limits, large plasma sample volumes (0.2–1 mL) and sample injection volumes are often required.2-6 These volumes are often impractical in discovery studies. Thus, the demand for quantitative bioanalytical assays that use decreased sample volumes, while maintaining or improving sensitivity are highly desired.
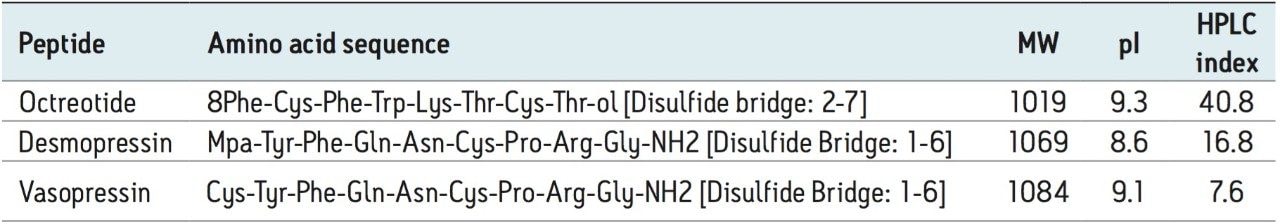
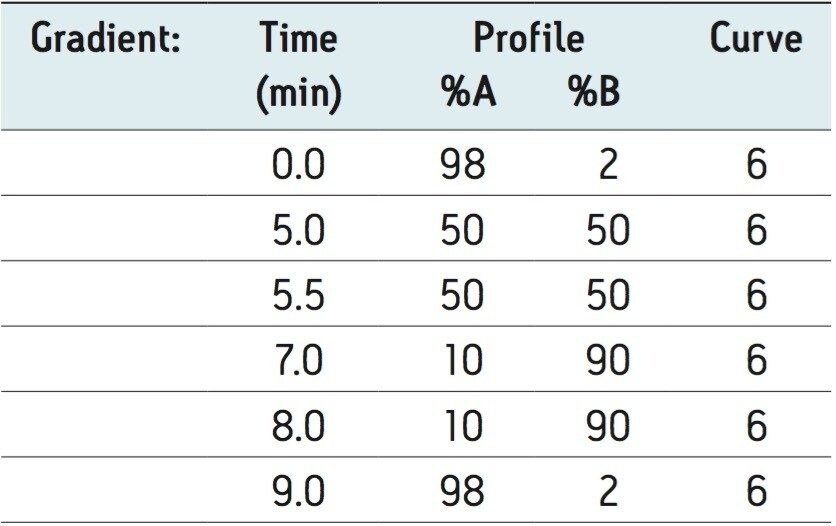
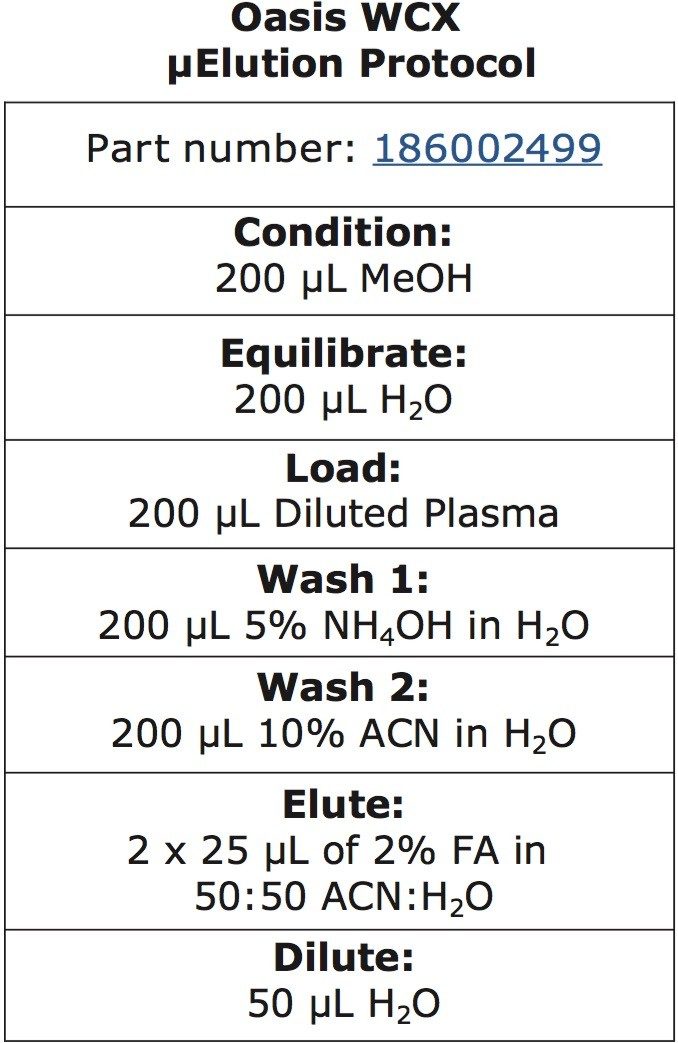
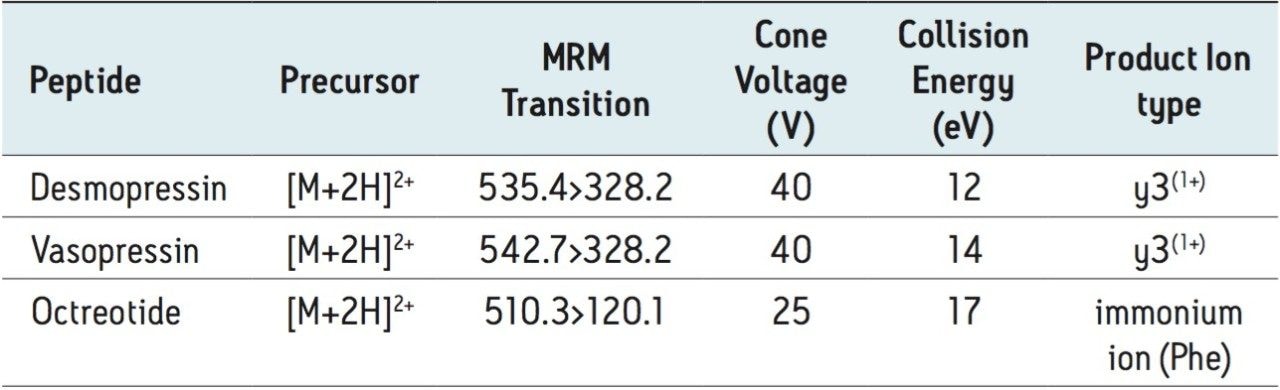
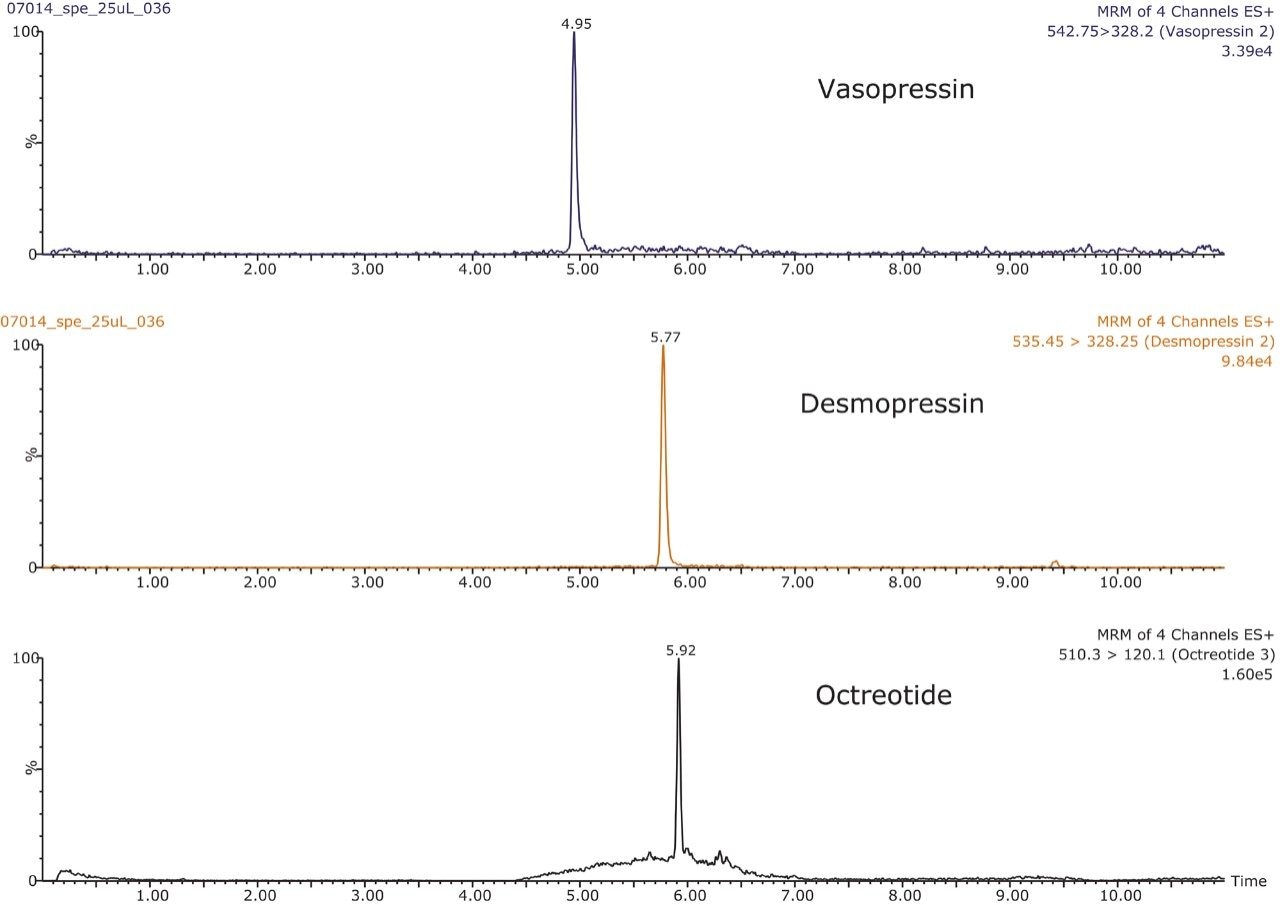
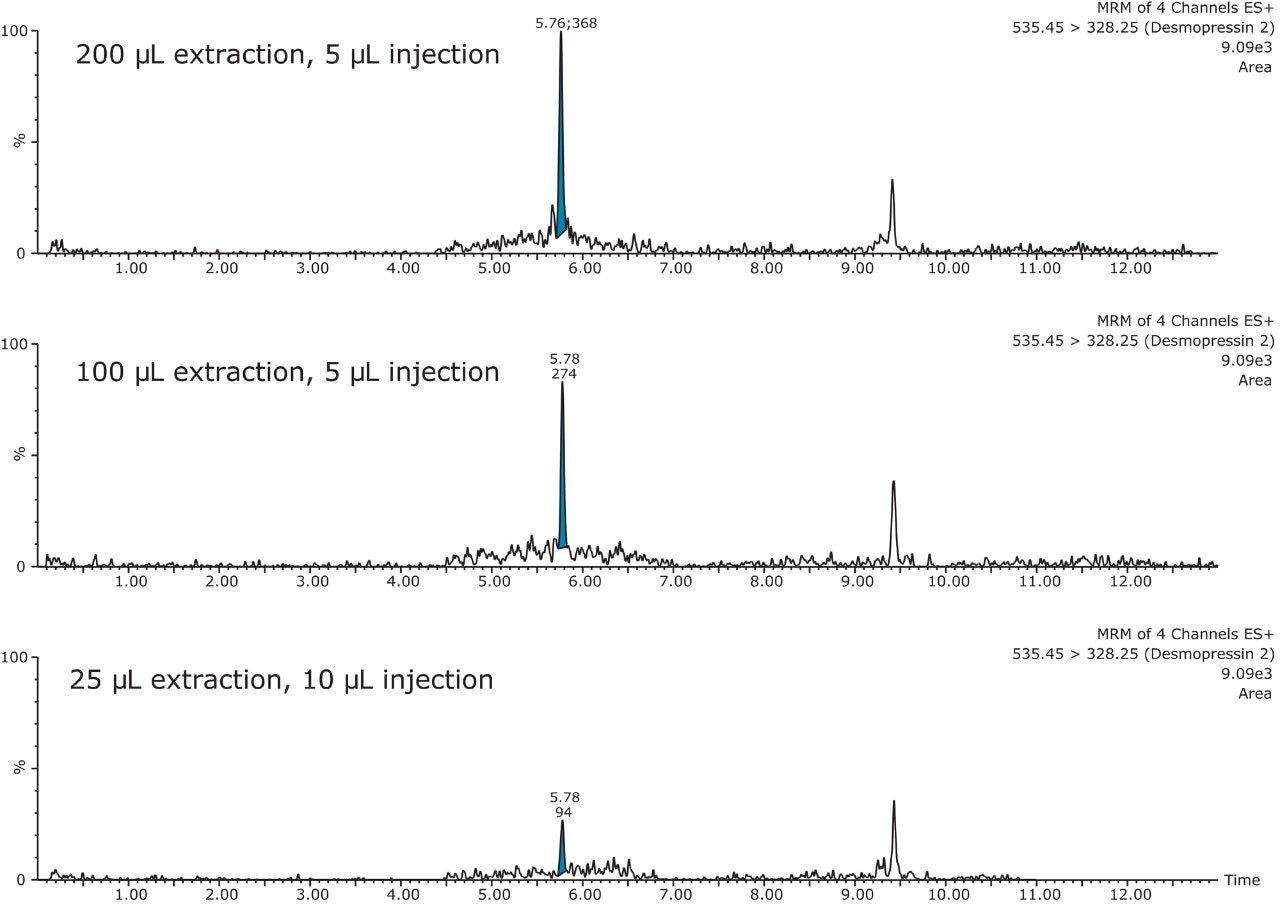
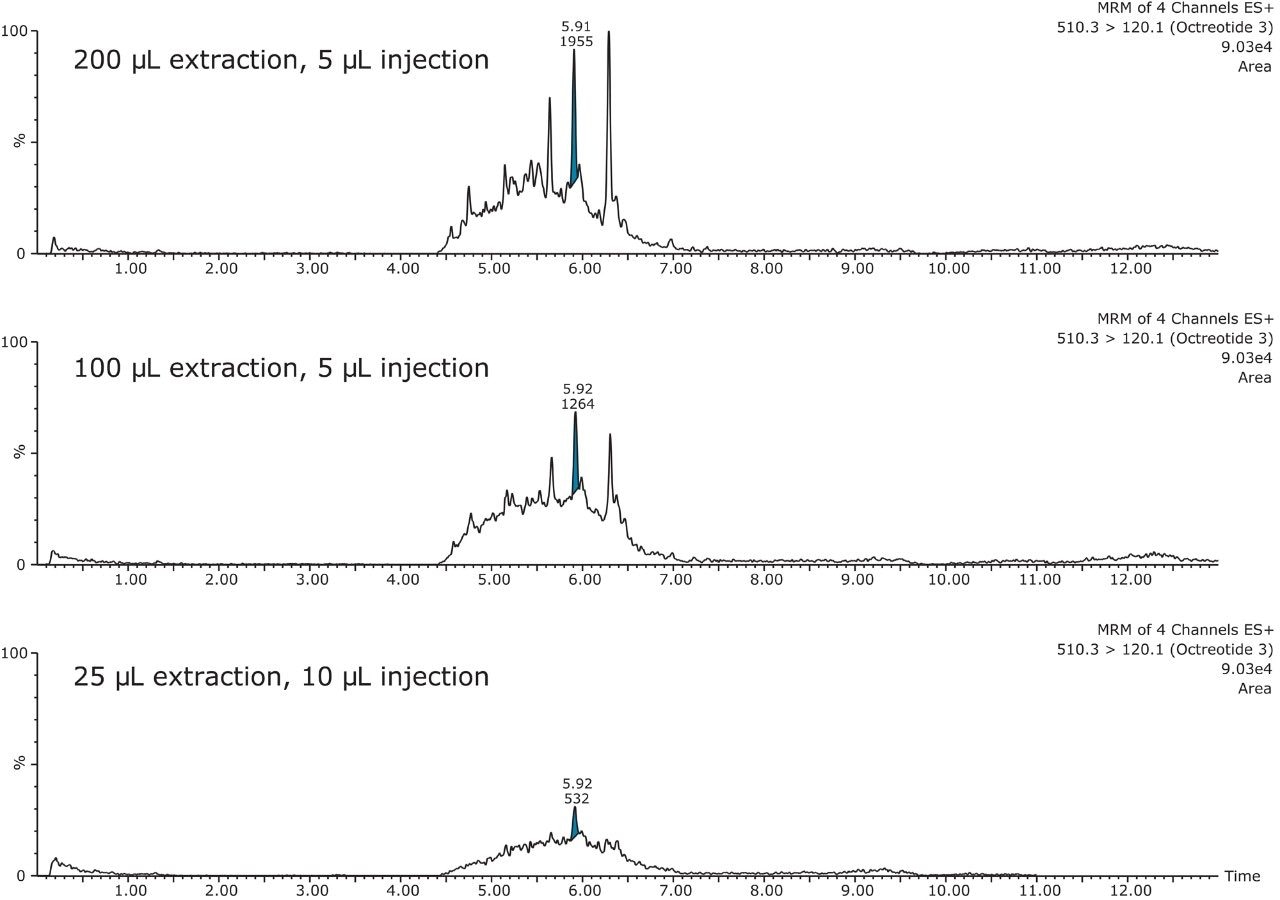
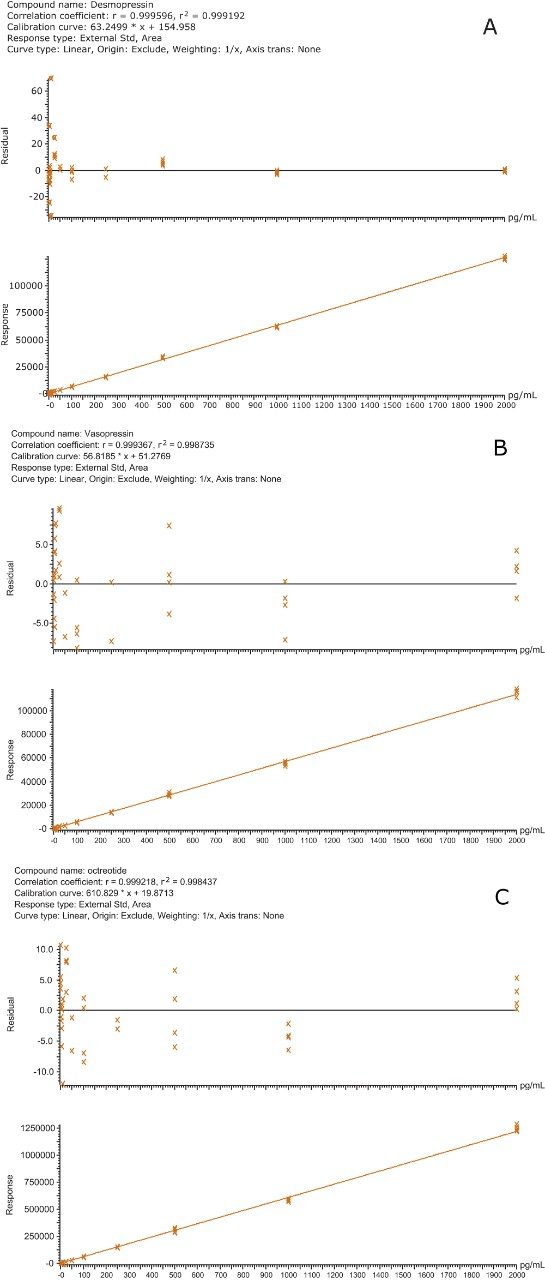
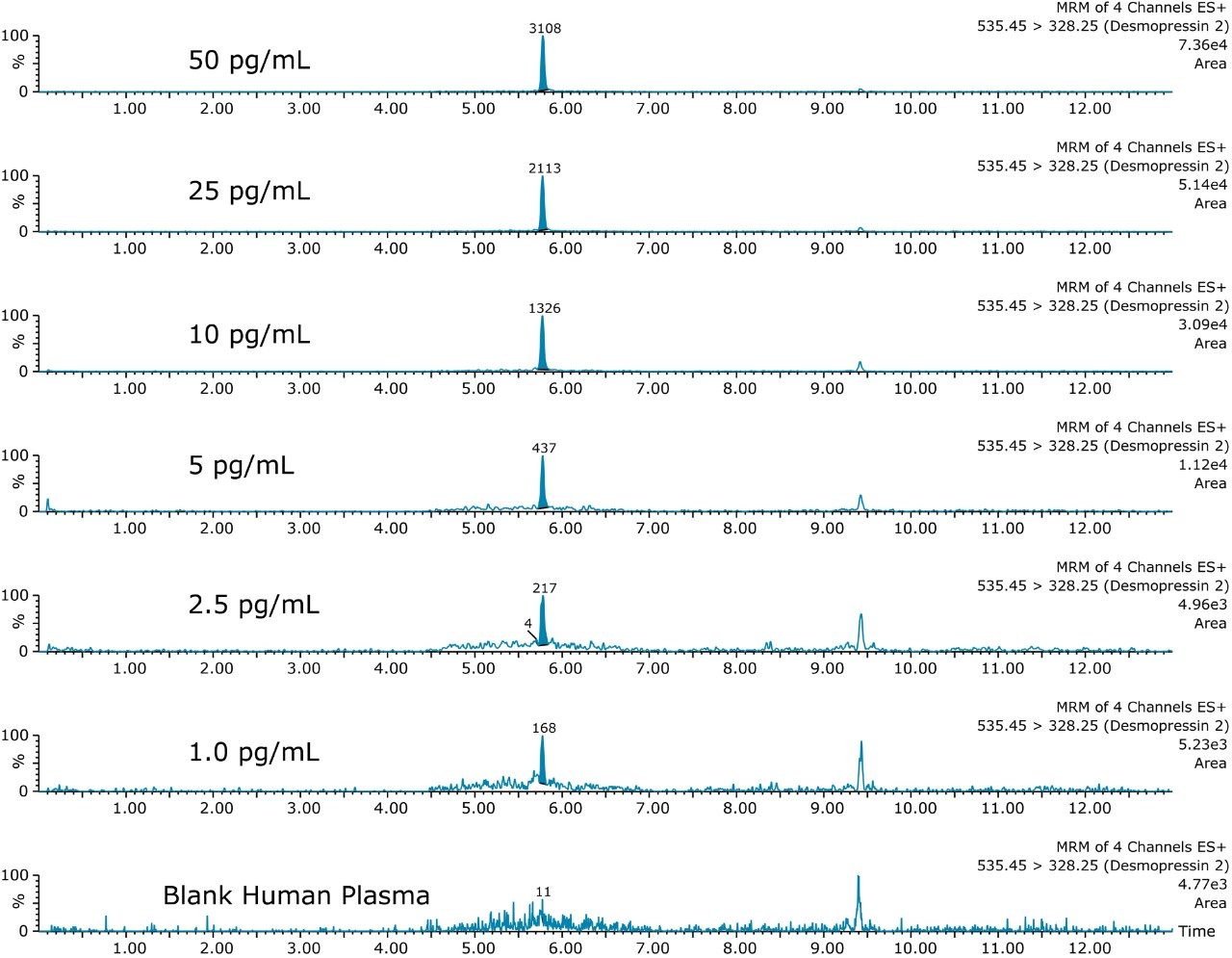
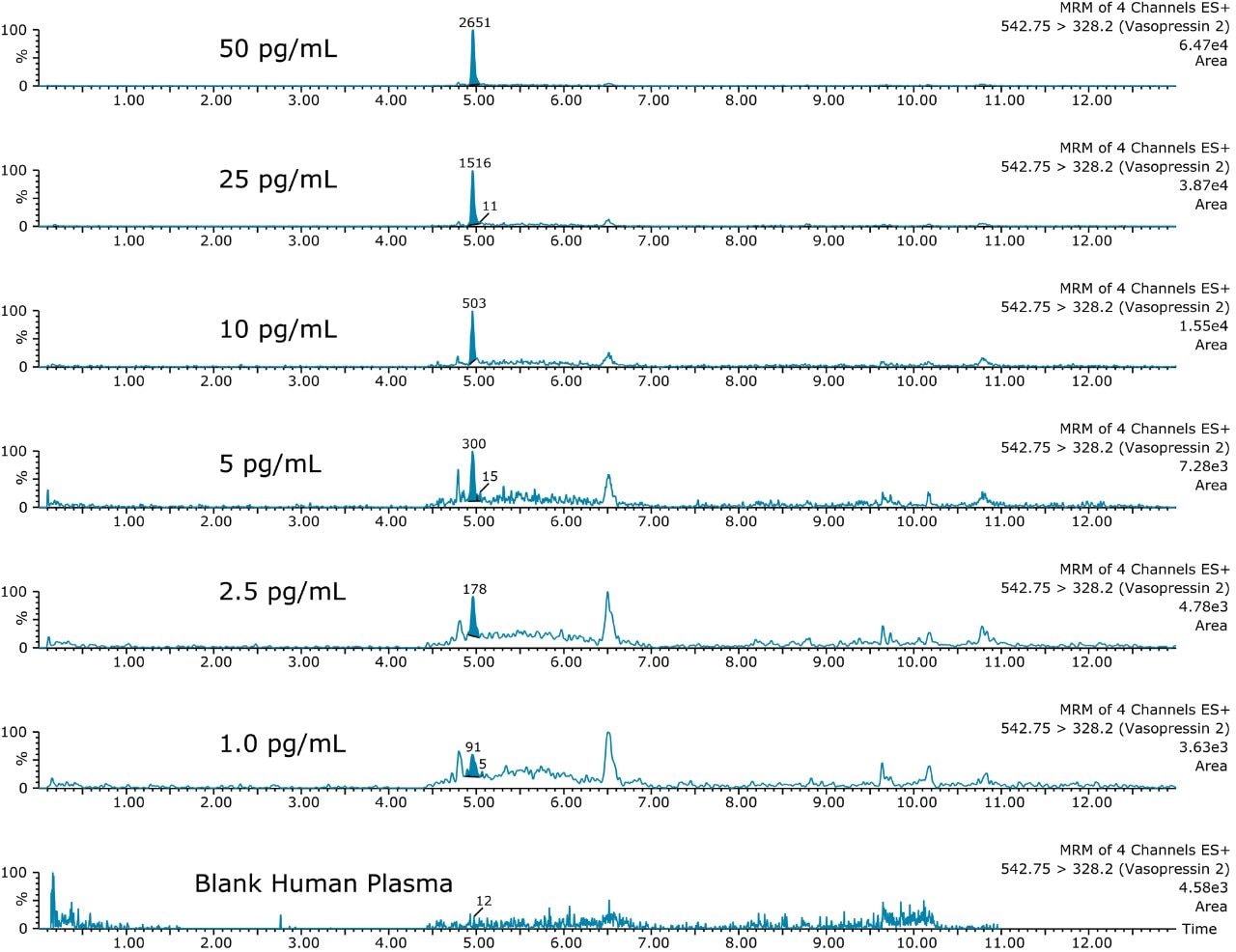
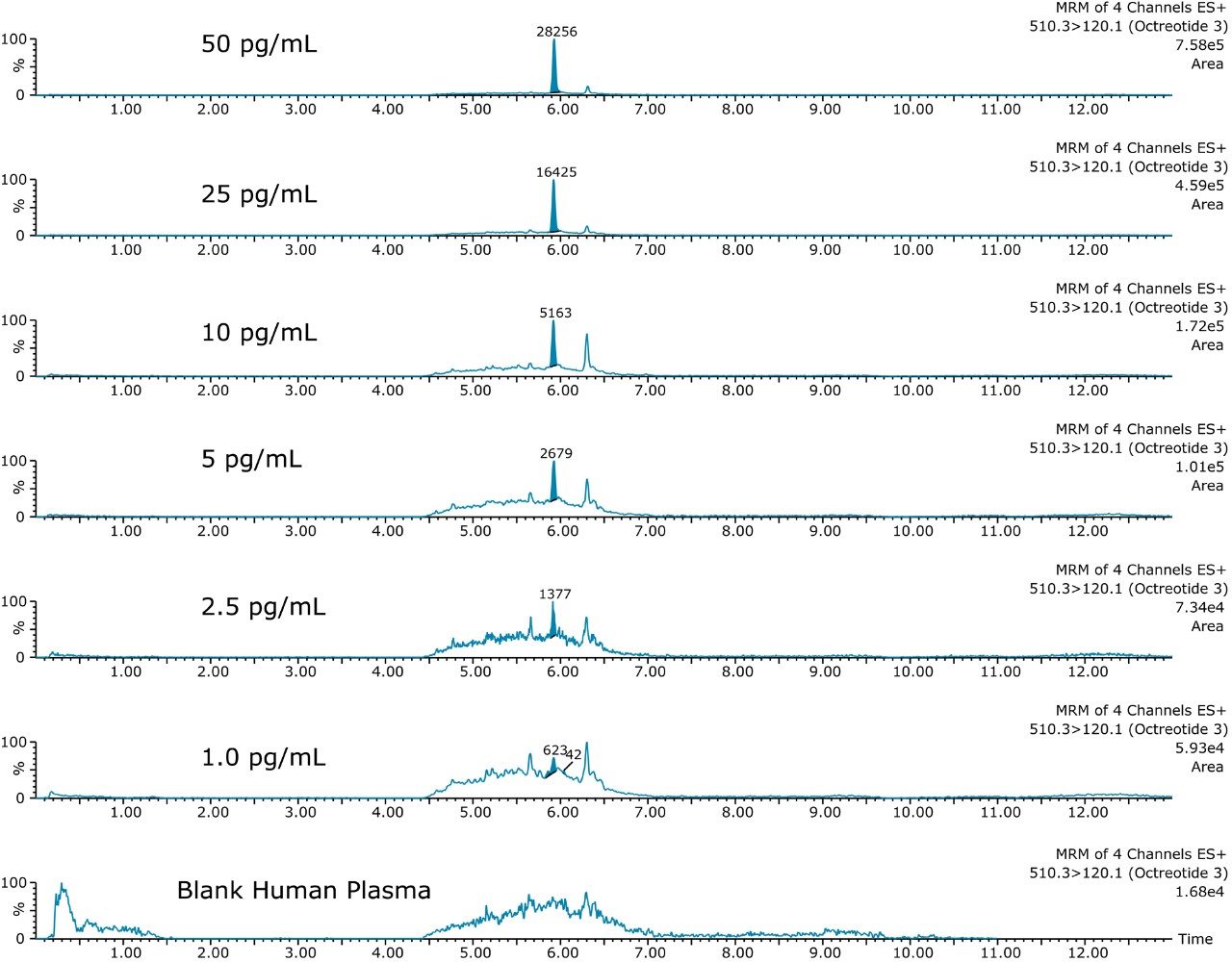
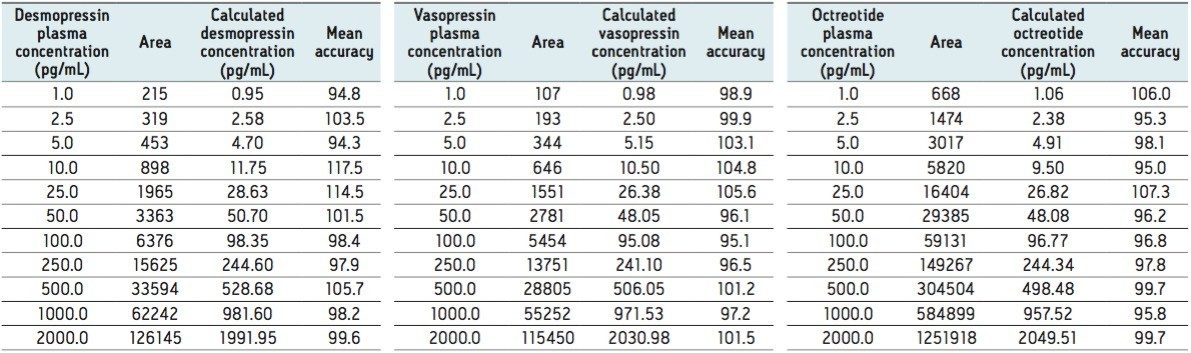
This application investigates the improved sensitivity and decreased sample volume requirements for the therapeutic and endogenous cyclic peptides: desmopressin, vasopressin and octreotide.7-9 The general properties of these peptides are shown in Table 1. Using a combination of selective μElution mixed-mode SPE sample preparation, optimal MS precursor and fragment choice, and the ionKey/MS System (source shown in Figure 1), limits of quantification of 1 pg/mL in plasma were achieved. Capitalizing on the attributes of the ionKey/MS System facilitated reducing plasma sample required to 25–100 μL.