Residual host cell proteins (HCPs) are low-level (1–100 ppm) process-related impurities that might be present in protein biopharmaceuticals even after extensive purification. HCPs could produce unwanted immunogenic responses in patients, they can reduce the efficacy or the stability of the drug, or they can be responsible for drug degradation. For these reasons, the regulatory agencies required that HCPs are identified and quantified prior to drug approval. The biopharmaceutical industry relies on ELISA assays for measuring the total HCP concentration expressed in ppm (or ng HCPs/mg biopharmaceutical). Mass spectrometry based HCP analysis has emerged in recent years as a powerful alternative to ELISA because it provides more extensive (proteome-wide) HCP coverage and is able to measure individual HCP levels.1-3
HCPs are identified using LC-MS either via data-dependent (DDA) or by data-independent (DIA) acquisition. In comparison with DIA, the DDA acquisition methods are less reproducible and cannot provide adequate HCP quantification because most of the instrument time is spent in MS/MS mode. In this application note, DIA and MSE acronyms are used interchangeably. In MSE acquisition, an equal amount of instrument time is spent in both MS and MS/MS modes using an alternate scanning mode with low and high collision energy switching.
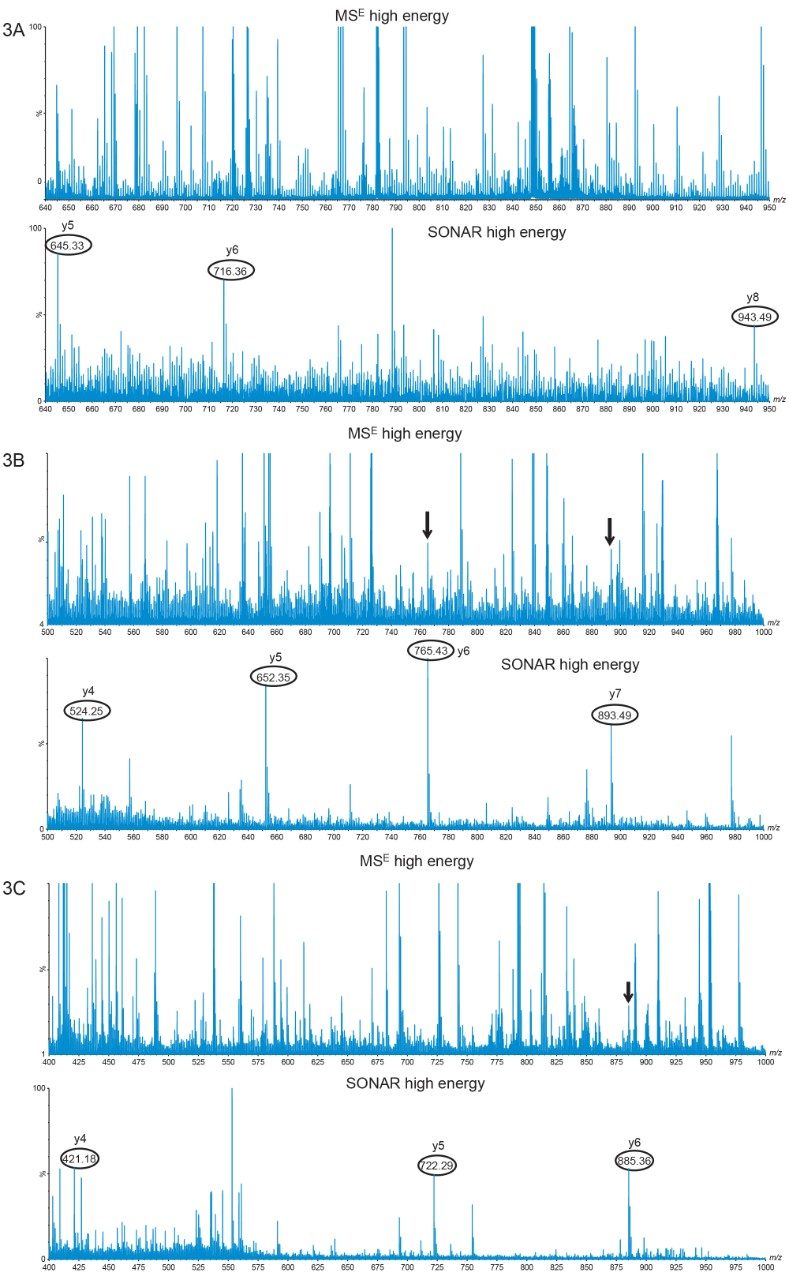
Here we investigated the capabilities of a novel data independent acquisition mode recently implemented on a Waters Quadrupole/Time-of-Flight (QTof) Mass Spectrometer, namely SONAR data acquisition. Instead of transmitting all peptide ions produced by electrospray ionization, in SONAR mode, the quadrupole scans over the mass range of interest during the time required for recording a single mass spectrum by the ToF analyzer. Co-eluting precursor ions with different m/z are separated during the rapid quadrupole scan and their corresponding fragmentation spectra are acquired using an identical quadrupole separation. In this way, SONAR offers additional selectivity, by producing cleaner MS/MS spectra with less interference. Two applications of this novel technology, in the area of lipid profiling,4 and Proteomics,5 have been recently described.
The first step of the HCP identification and quantification workflow described here and presented in Figure 1 is based on the HCP Discovery Assay performed in SONAR mode using extensive (90 min) peptide separations. Following data processing with Progenesis QI for Proteomics 4.0, the HCPs are identified by a proteome-wide database search. In addition, SONAR MS/MS fragmentation spectra can be assembled into spectral libraries, containing peptide precursors, charge states, and retention times. In the second step of the HCP workflow, additional HCP samples resulted from the purification of the same biopharmaceutical, are analyzed by higher–throughput HCP monitoring assays employing MSE data acquisition with 30 min peptide separations. The LC-MSE dataset is then searched against the spectral library for HCP quantification and monitoring at every step during biopharmaceutical purification. The HCP workflow described in Figure 1 was applied for identification and monitoring of HCPs from the NIST monoclonal antibody.