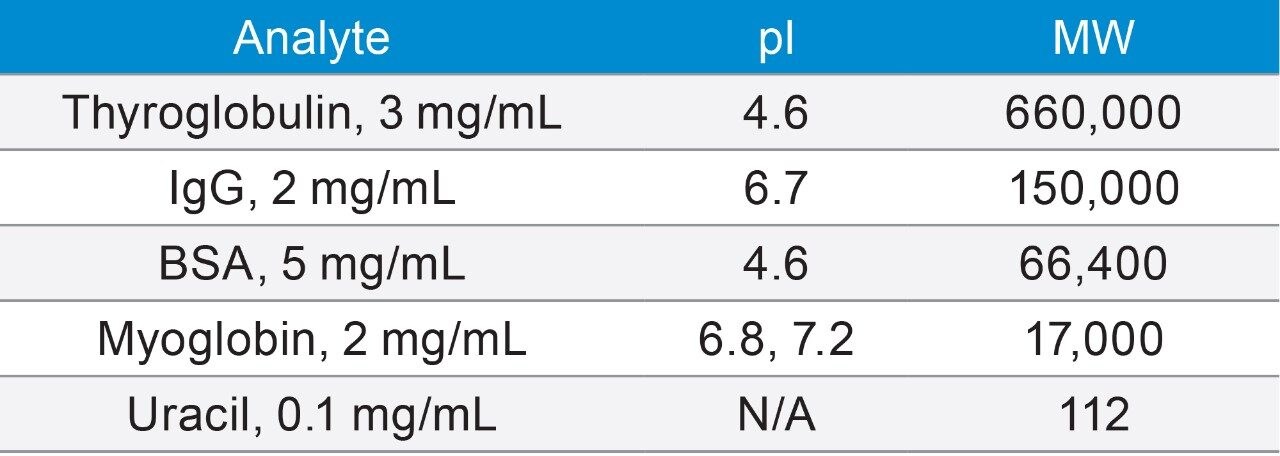
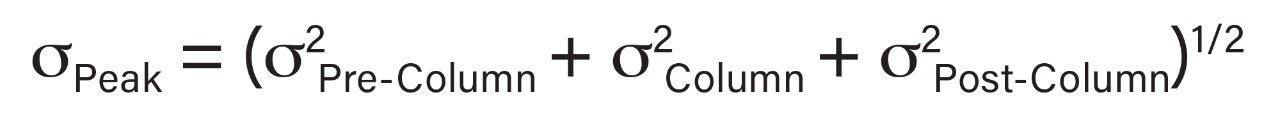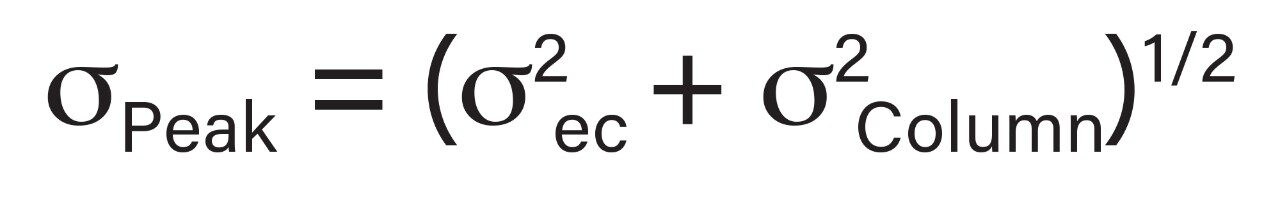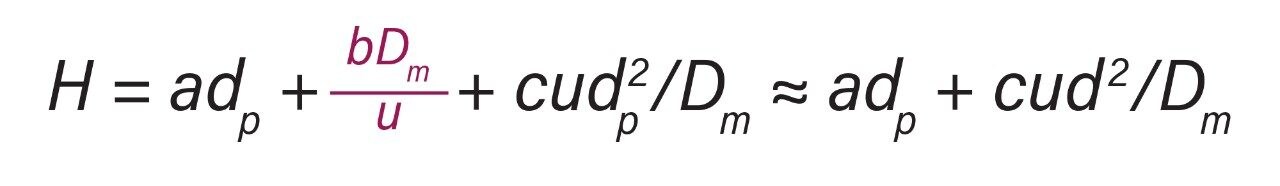Impact of System Dispersion on SEC Separation Efficiency
It can be assumed that in the absence of extra-column dispersion, if two columns of the same length, but different internal diameters, are packed with the same particles and with the same plate count, then those columns will provide the same resolution when the same linear velocity, and a proportional sample load relative to the column volume, are maintained. Or more simply stated, resolution is independent of column I.D. Under these constraints, the peak widths observed for these two different size columns will be equivalent in the time domain, but the volume of a peak for the larger I.D. column will be proportionally larger due to its higher flow rate. The practical implications of these relationships are that as SEC peak volumes become smaller due to the use of smaller internal diameter columns, extra-column dispersion can have a greater deleterious impact on the net chromatographic result.
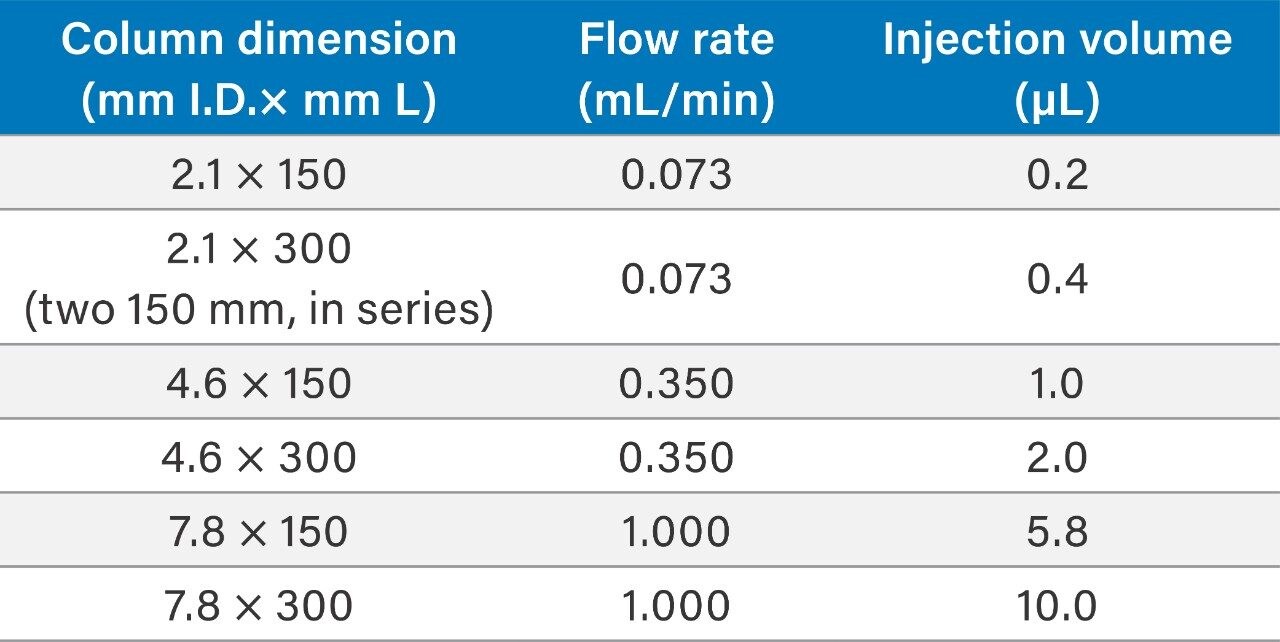
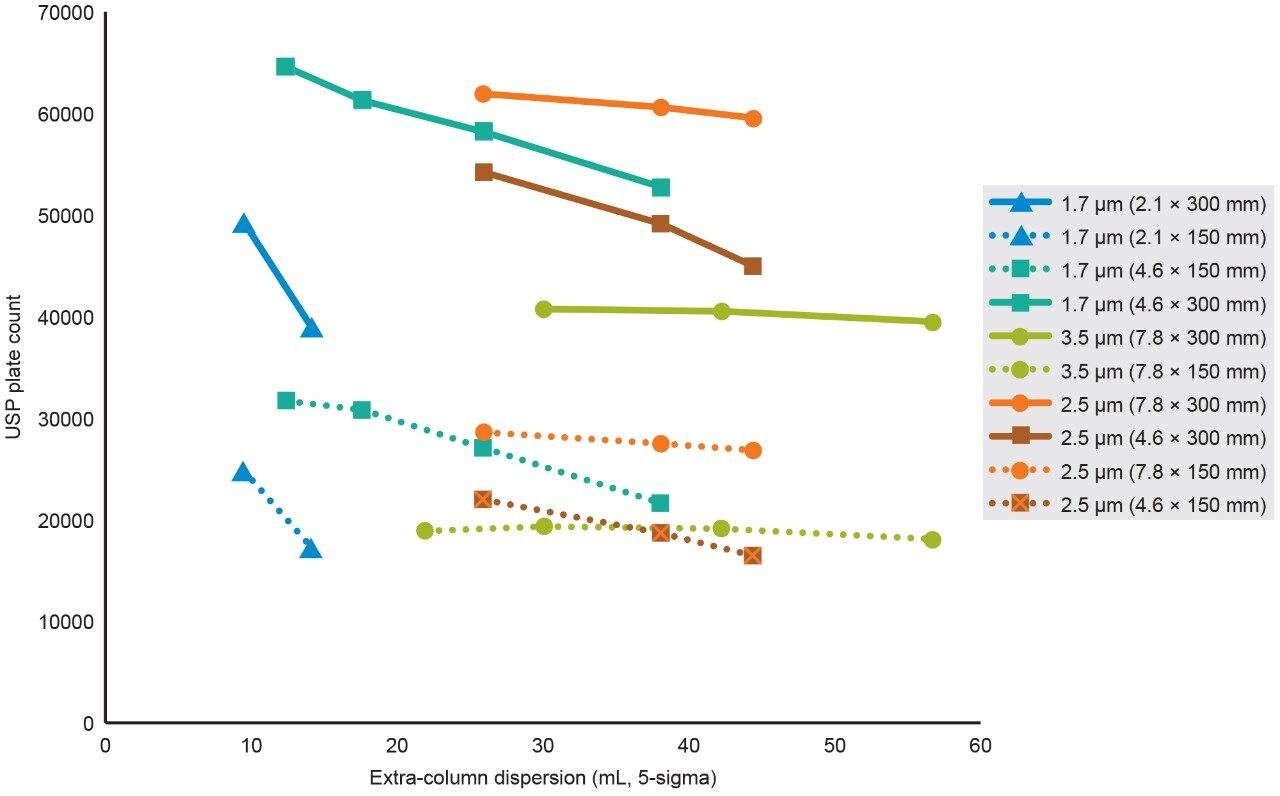
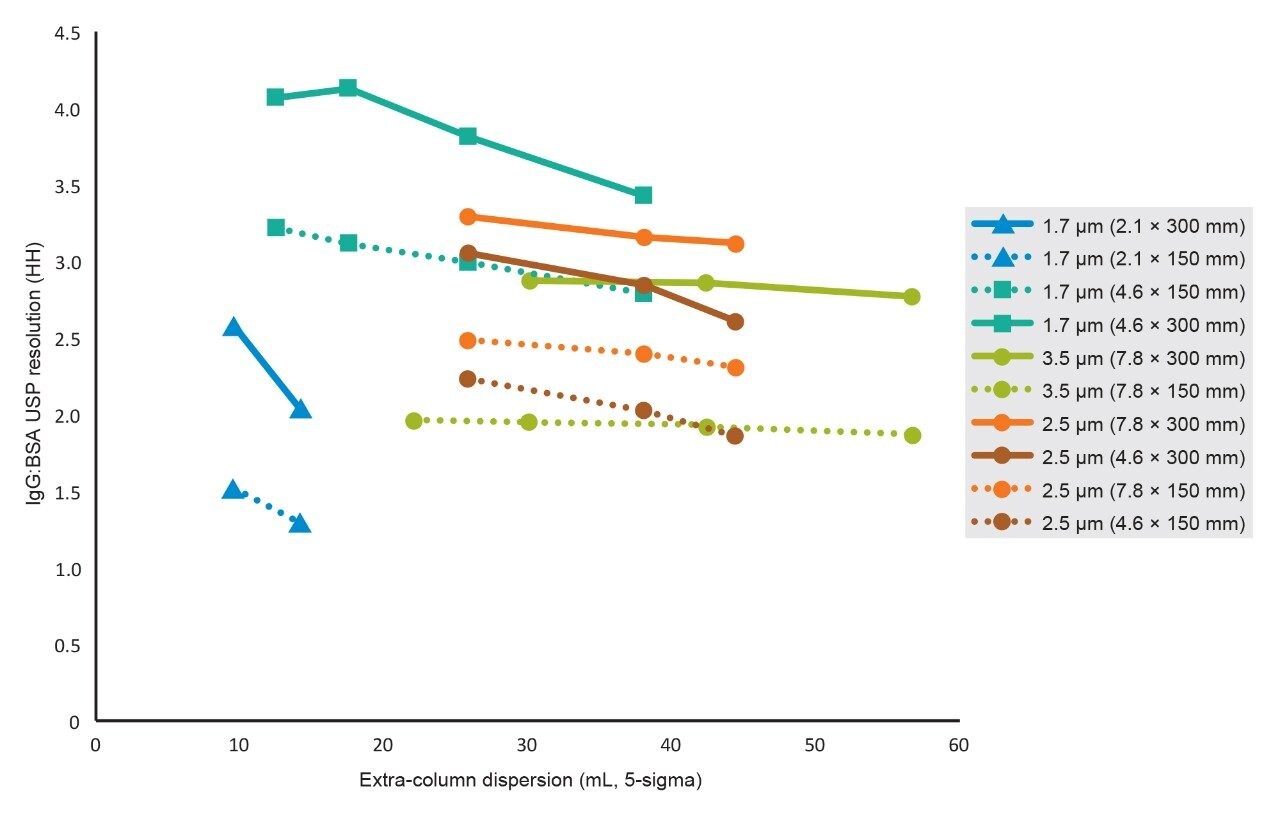
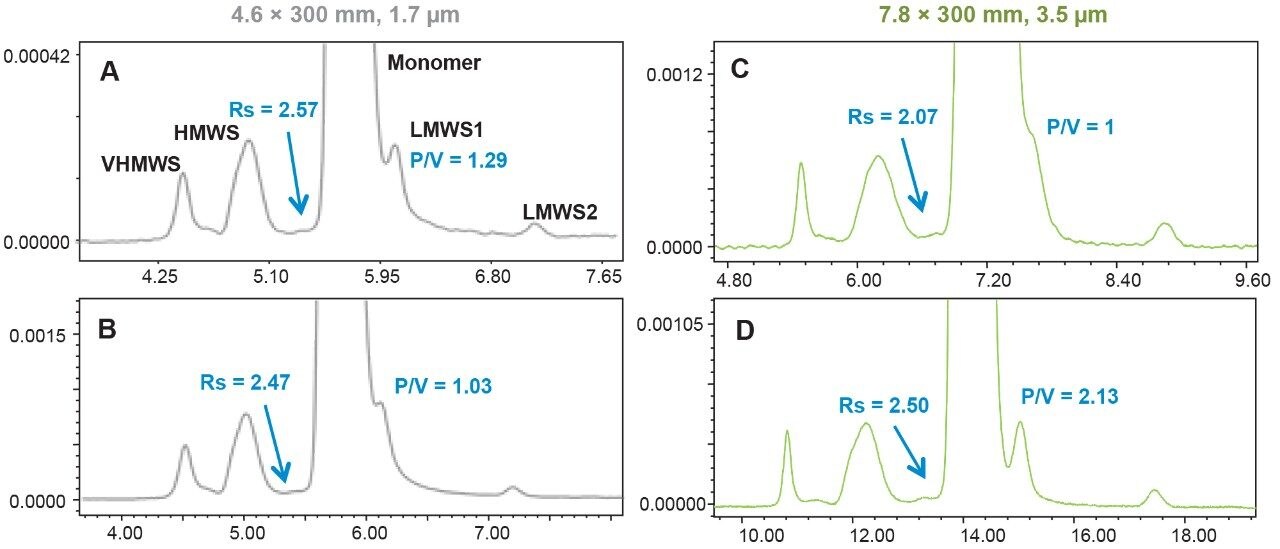
To assess the impact that σec has on SEC separations as a function of particle size, column I.D., and column length, the SEC MW protein standard mix was injected on 2.1 mm and 4.6 mm I.D. columns packed with 1.7 μm particles, 4.6 mm and 7.8 mm I.D. columns were packed with 2.5 μm particles, and 7.8 mm I.D. columns were packed with 3.5 μm particles. Column lengths of 150 and 300 mm were tested, and, in all cases, the average pore diameter of the particles was 200 Å. The 300 mm bed length, 2.1 mm I.D. column was simulated by running two 2.1 × 150 mm length columns, in series. For these experiments, the linear velocities were held constant, therefore, for a given column length the analysis times are equivalent. In addition, sample loads were appropriately scaled based on column volume.
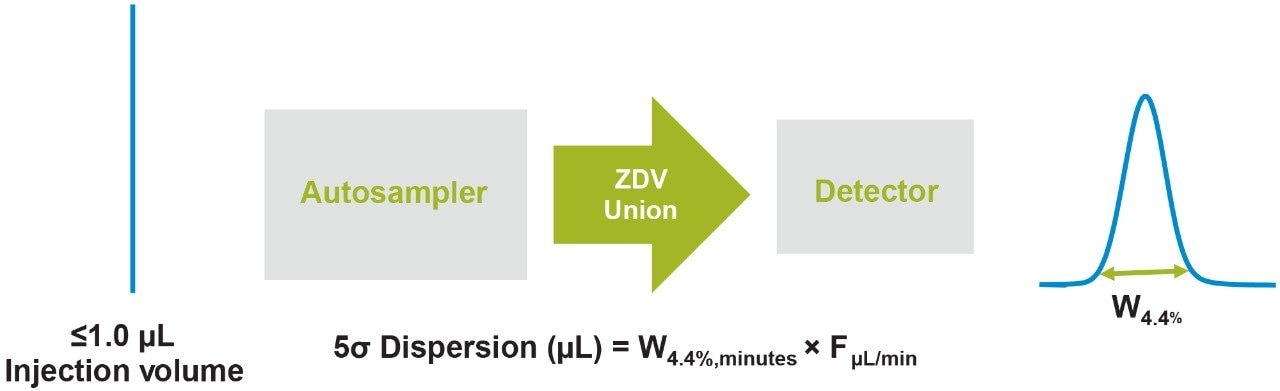
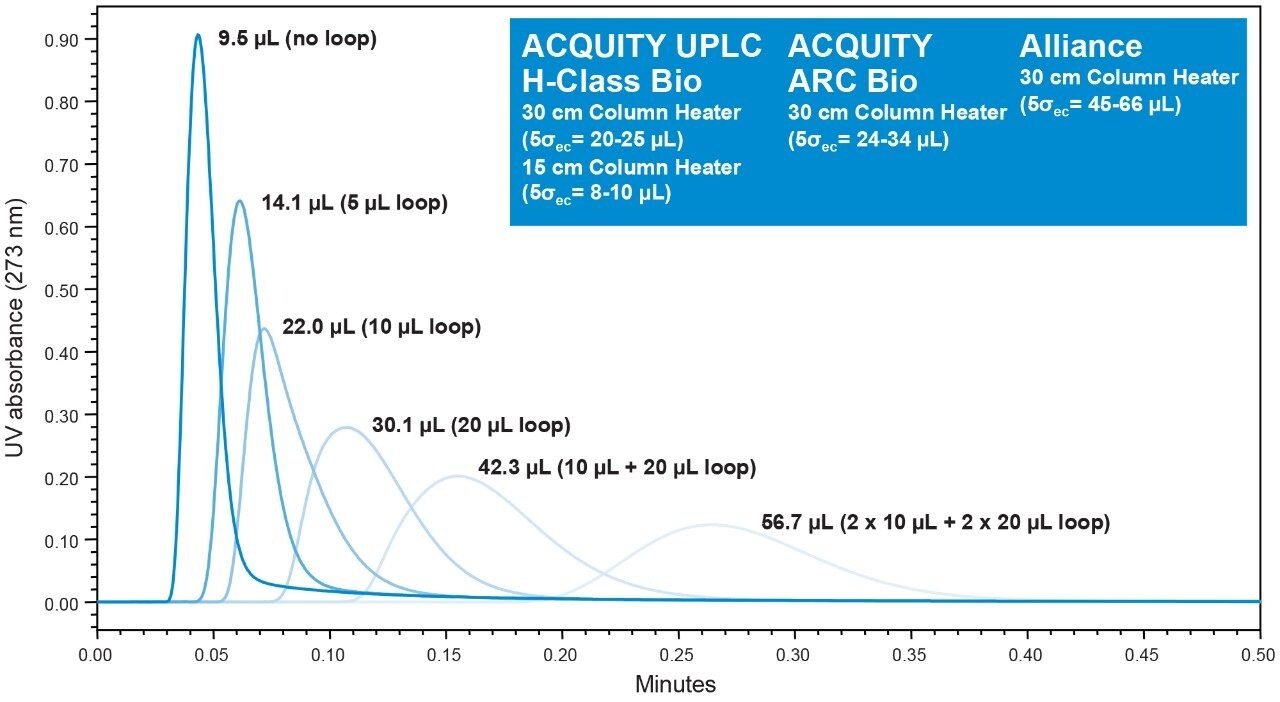
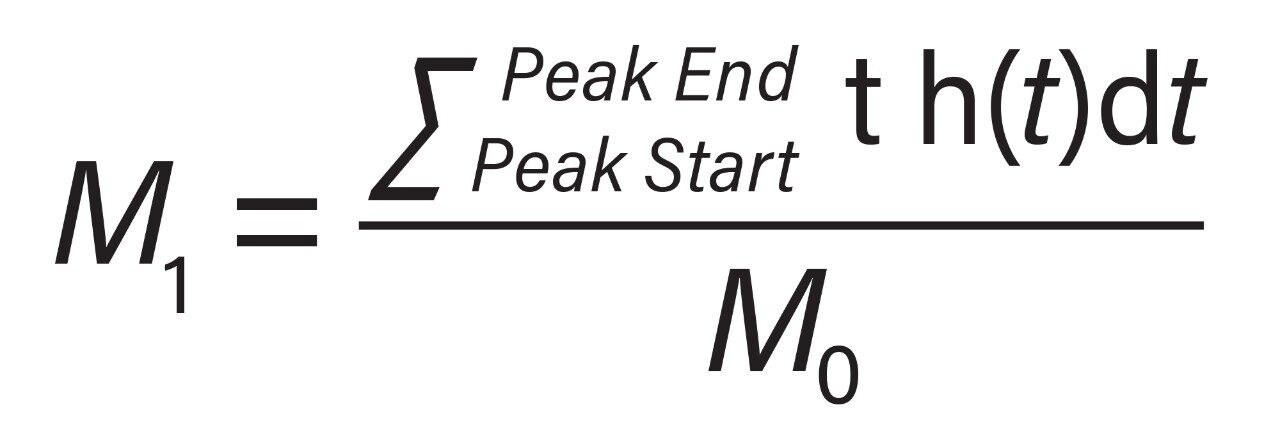
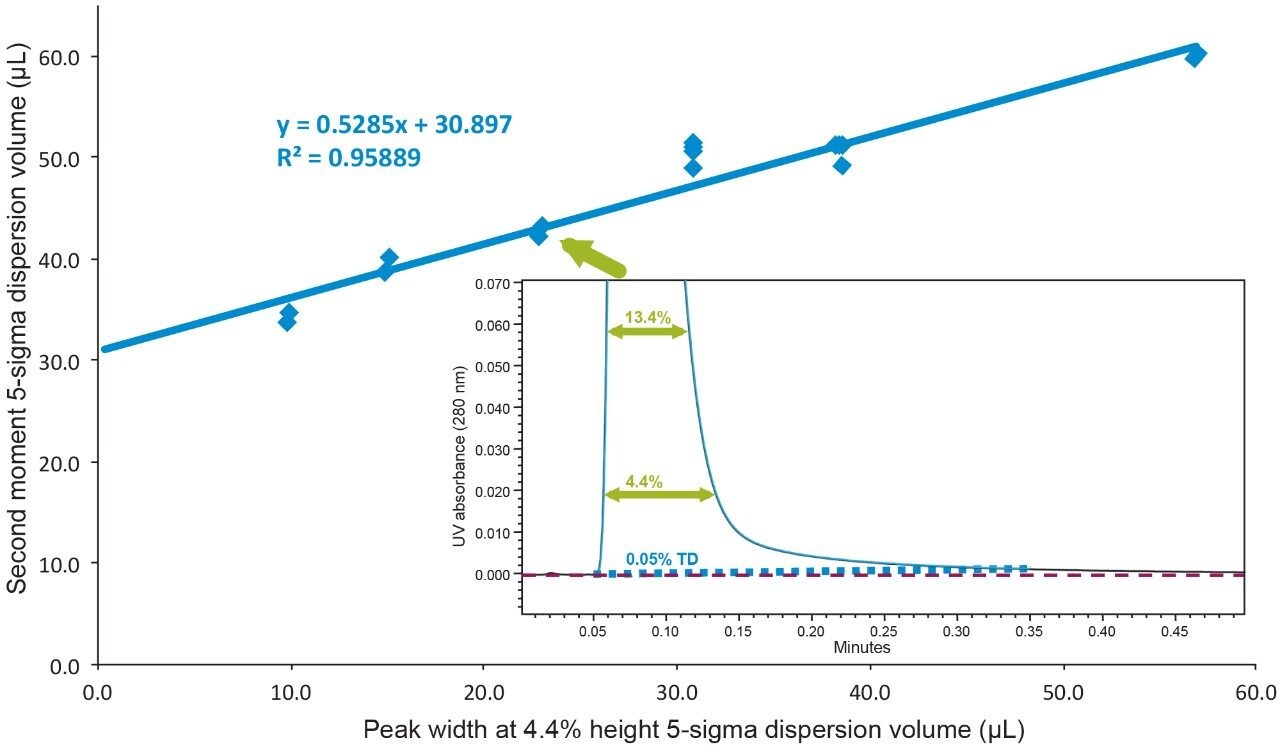
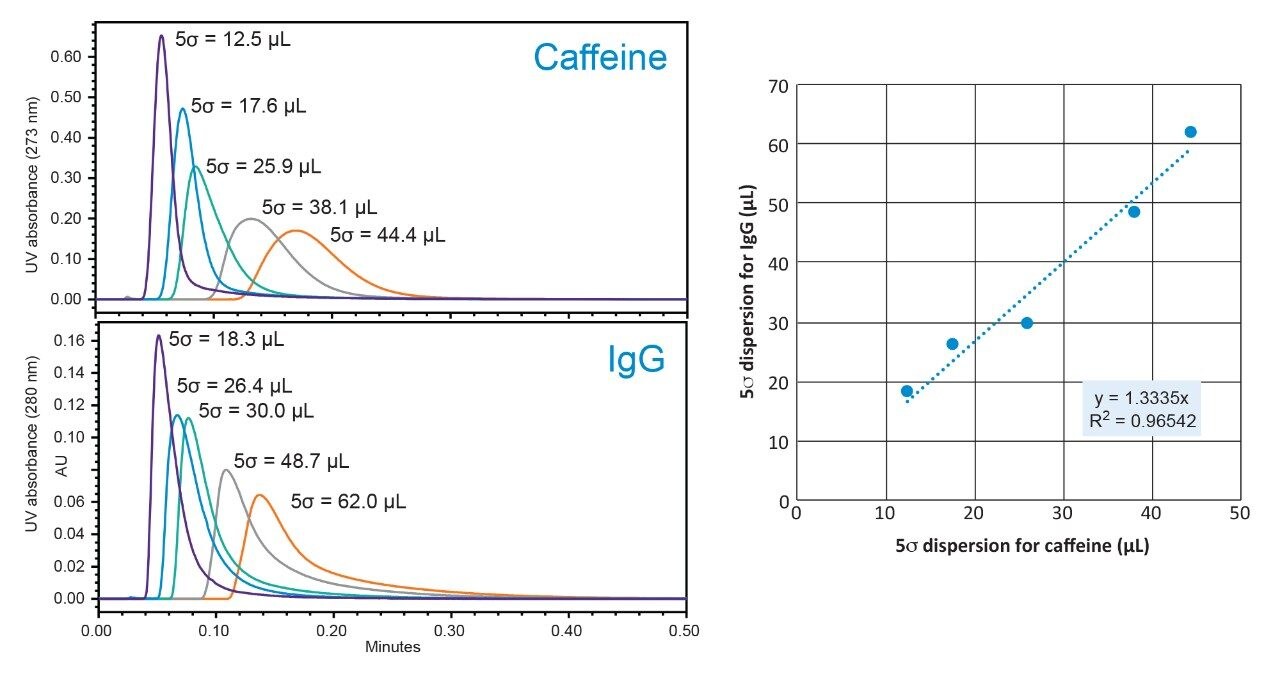
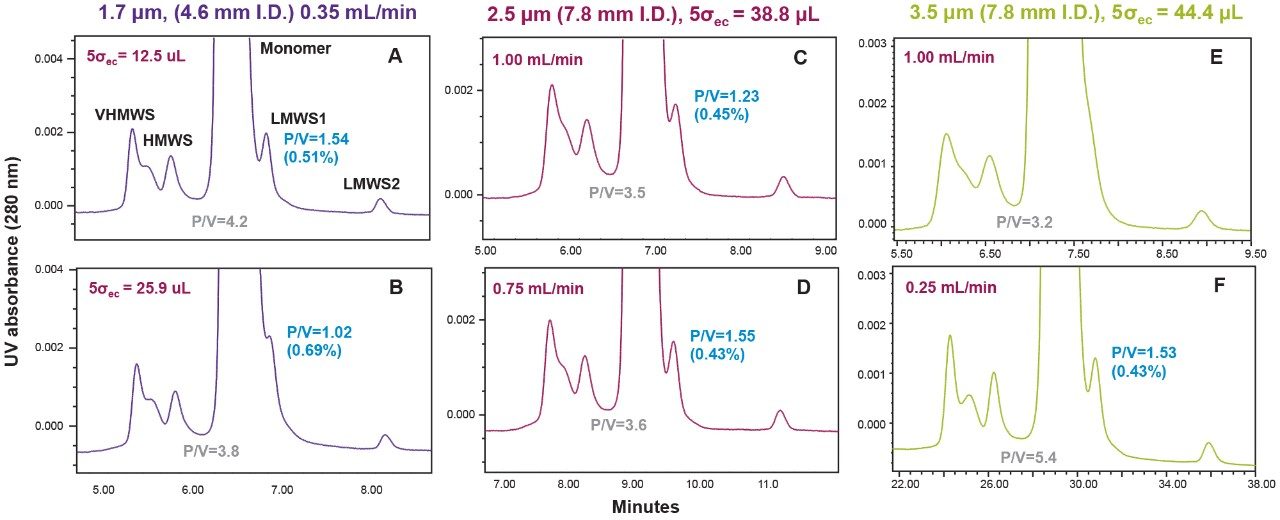
Representative chromatographic profiles for the 2.1 mm and 4.6 mm I.D. columns packed with 1.7 μm particles obtained over a range of system dispersions, are presented in Figure 5. These chromatograms were selected as they provide the most visually discernable changes. Quantitative measures of the USP plate counts (based on uracil) and the resolution between the two largest baseline resolved proteins, IgG and BSA, are presented in Figures 6 and 7, respectively. Additionally, to get a sense of the peak volumes produced by these SEC columns, we determined the estimated 5σec peak volumes for the proteins IgG, BSA, and myoglobin based on the direct measurement of peak width at 4.4% peak height (Figure 8). To obtain a better estimate of the 5σcolumn volumes, we have subtracted the contributed 5σec from the measured protein peak widths (5σpeak) using the relationship shown in Equation 1.