Apiculture relies on antibiotics to prevent disease propagating through the densely populated bee colonies. The overuse of antibiotics in honey bee colonies can cause high levels of residues in honey products, which becomes a public health issue. Additionally, bacteria that have developed resistance to the applied antibiotics can pose an increased threat to both human and animal health. Consequently, antibiotics become less effective against bacteria and there will be fewer alternatives available for the successful treatment of infection. Unscrupulous producers search for these alternative antibiotics such as nitrofurans to treat disease.
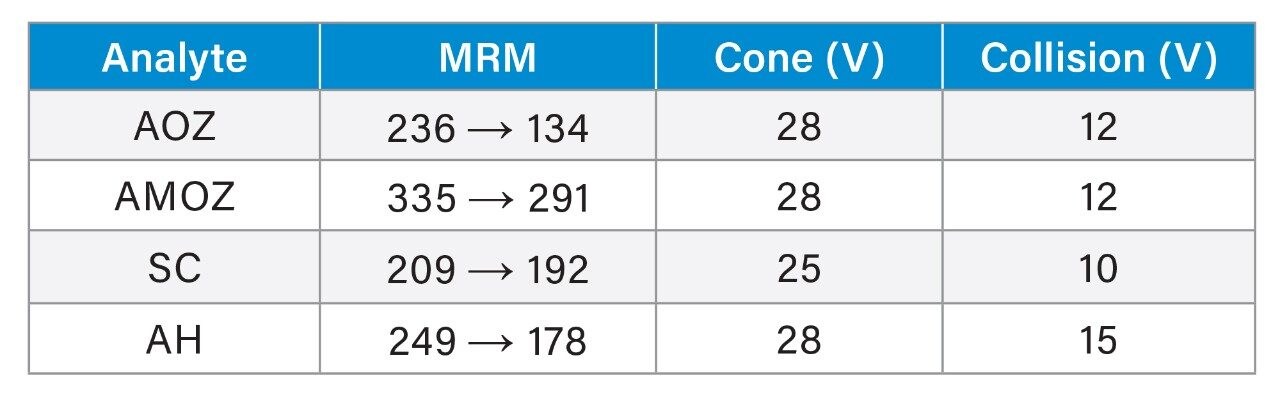
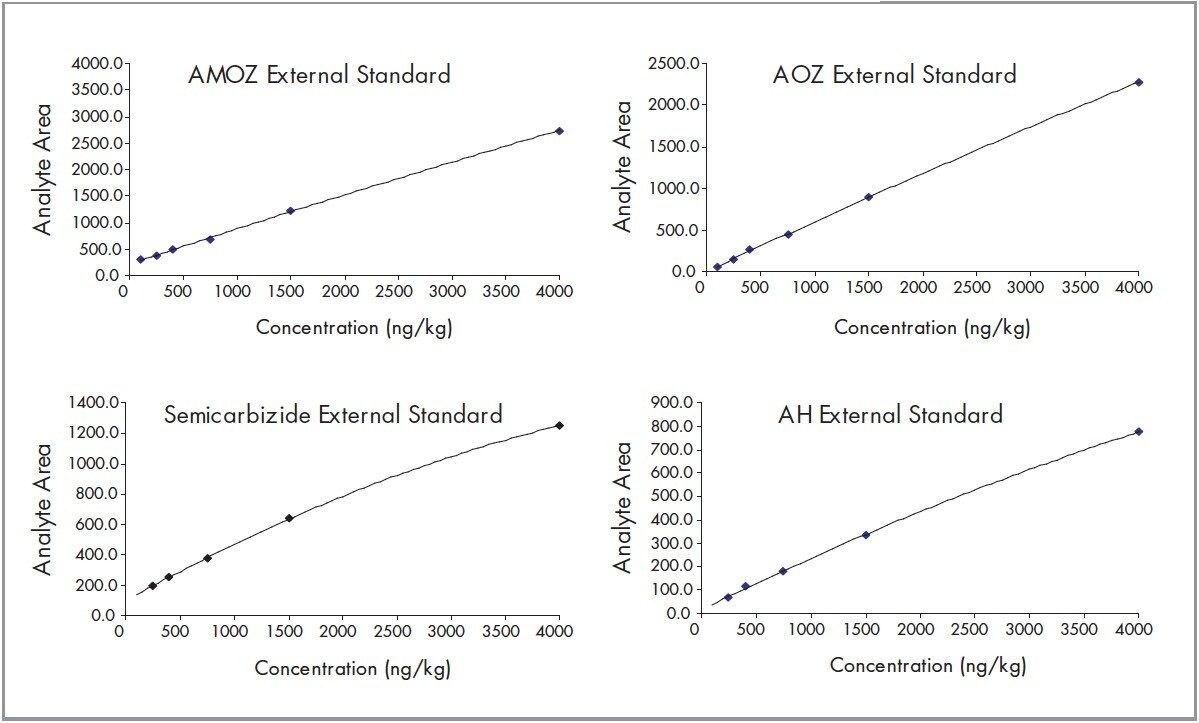
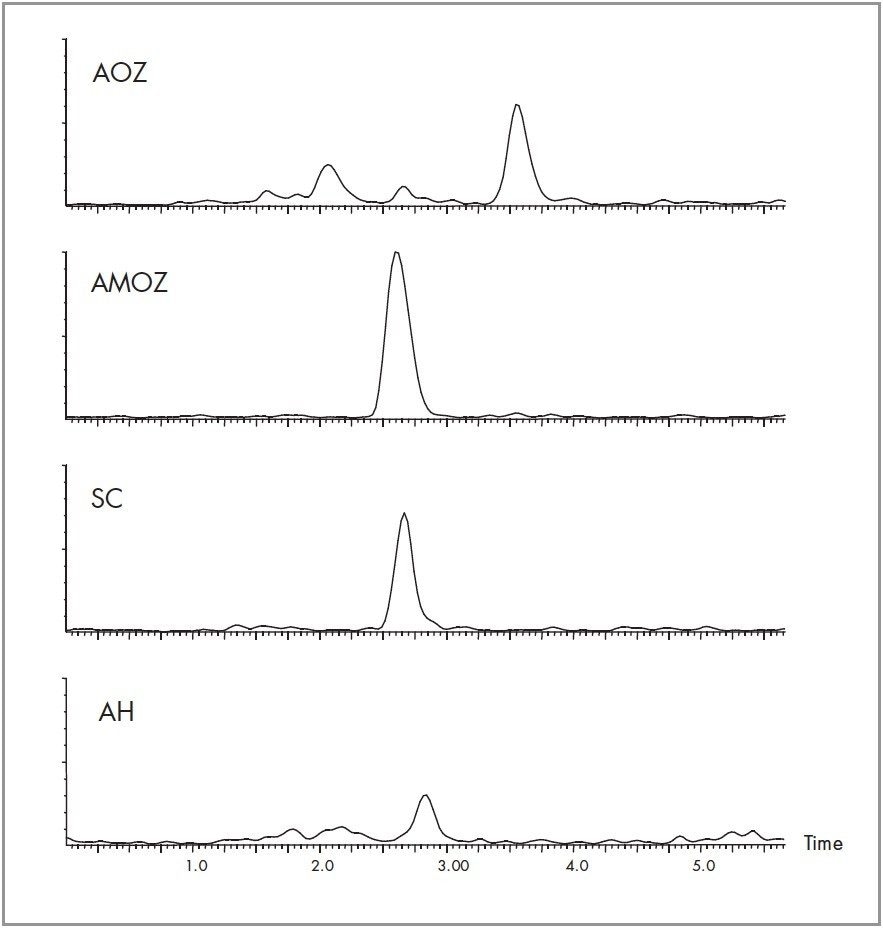
Nitrofuran antibiotics metabolize rapidly with an in vivo half-life in the order of hours, making parent drug detection ineffective. An LC-MS/MS method is described for the quantification of nitrofuran metabolite residues (AOZ, AMOZ, 1-Aminohydantion(AH), Semicarbizide (SC)) in commercially available honey. The metabolite residues were extracted from the honey samples by first dissolving the honey in HCl. The samples were cleaned, derivatized and then enriched using Oasis HLB solid phase extraction devices. The metabolite residues were resolved chromatographically using a XTerra MS C18 analytical Column. Positive ion electrospray mass spectrometry was used to quantify and confirm the parent ion [M+H]+ and fragments for each target analyte.