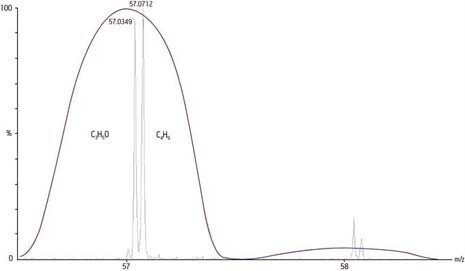
Confronto della precisione da strumento a strumento: unità in millimassa (mmu), errore di misura (ppm) e risoluzione
Secondo la Accurate Mass Best Practice Guide del programma VIMMS, un’iniziativa che fa parte del sistema di misurazione nazionale del Regno Unito, la maggior parte degli strumenti utilizzati per misurazioni di massa accurate è in grado di raggiungere una precisione pari o superiore a 10 ppm.
Una massa calcolata di 118 Da misurata da un moderno spettrometro di massa con un’accuratezza di 2 mmu mostrerebbe un errore di 17 ppm, sufficiente per gli standard odierni per la determinazione univoca di una formula chimica di tale massa:
Massa esatta calcolata monoisotopica = 118 Da
Massa accurata misurata = 118,002 Da
Differenza = 0,002 mmu
Errore [Differenza/massa esatta x 106] = 17 ppm
Uno strumento in grado di fornire una risposta a 750 m/z,, anch’essa carente di 2 mmu, avrebbe una precisione pari a 2,7 ppm. Nel primo caso, secondo gli standard pubblicati dal Journal of The American Society for Mass Spectrometry, la misura è più che sufficiente per l’identificazione univoca di una formula chimica. Ma, in quest’ultimo caso, la misurazione non è sufficientemente precisa. Solo la spettrometria di massa a risonanza ionica ciclotronica a trasformata di Fourier (FTICR) di ordine più alto può raggiungere tale precisione a masse più elevate.
Un metodo esaustivo per la valutazione della capacità di misurazione di massa accurata degli strumenti che richiama l’uso previsto è il calcolo del valore quadratico medio o errore RMS. Per illustrarne l’utilizzo, quanto segue è un adattamento delle specifiche di accuratezza di misura di massa di uno spettrometro di massa ToF in commercio.
“L’accuratezza della misurazione della massa dello strumento, in normali condizioni operative, sarà migliore di un dato valore ppm RMS su un intervallo m/z dato, in base a un numero di misurazioni ripetute consecutive di un picco dell’analita (di un dato valore m/z), utilizzando un picco di riferimento adatta (di un dato valore m/z). I picchi dell’analita e di riferimento devono avere un’intensità sufficiente ed essere privi di interferenze da parte di altre masse.”
Ci sono alcuni punti importanti e presupposti da considerare:
- È già stata eseguita una calibrazione dello strumento con picchi di massa nota utilizzando uno standard di calibrazione. Il picco di riferimento viene utilizzato per tenere conto di eventuali variazioni della calibrazione dello strumento nel tempo e l’accuratezza della misurazione della massa viene determinata utilizzando il picco dell’analita.
- Normali condizioni operative: possono includere anche dettagli sulle condizioni cromatografiche (per le specifiche delle prestazioni LC-MS) ed eventuali condizioni operative MS correlate (per esempio risoluzione della massa, m/z di interesse o velocità di scansionamento spettrale).
- Intensità sufficiente: si presuppone che il conteggio ionico non sia dannoso per la caratterizzazione della precisione e dell’accuratezza (misurazione di massa) dello strumento in questione. Un numero insufficiente di ioni comporta scarse statistiche relative agli ioni e un numero eccessivo di ioni può portare alla saturazione del rivelatore; entrambe le cose si traducono in una variazione maggiore nella deviazione standard delle misurazioni ripetute e incidono negativamente sul calcolo dell’errore RMS (rilevante anche per la calibrazione dello strumento).
- Privi di interferenze: si presuppone che la misurazione della massa del picco di massa nota sia priva di interferenze da parte di ioni di massa identica o simile. La sovrapposizione dei picchi comporta una scarsa accuratezza delle misure di massa, che è anche dannosa per la caratterizzazione adeguata dell’accuratezza o della precisione dello strumento (rilevante anche per la calibrazione dello strumento).
- Il riferimento è una buona rappresentazione dell’intervallo m/z rilevante per l’analisi di un particolare tipo di campione.
L’errore RMS viene calcolato utilizzando la seguente relazione, in cui Eppm è l’errore ppm e n è il numero di masse considerate: