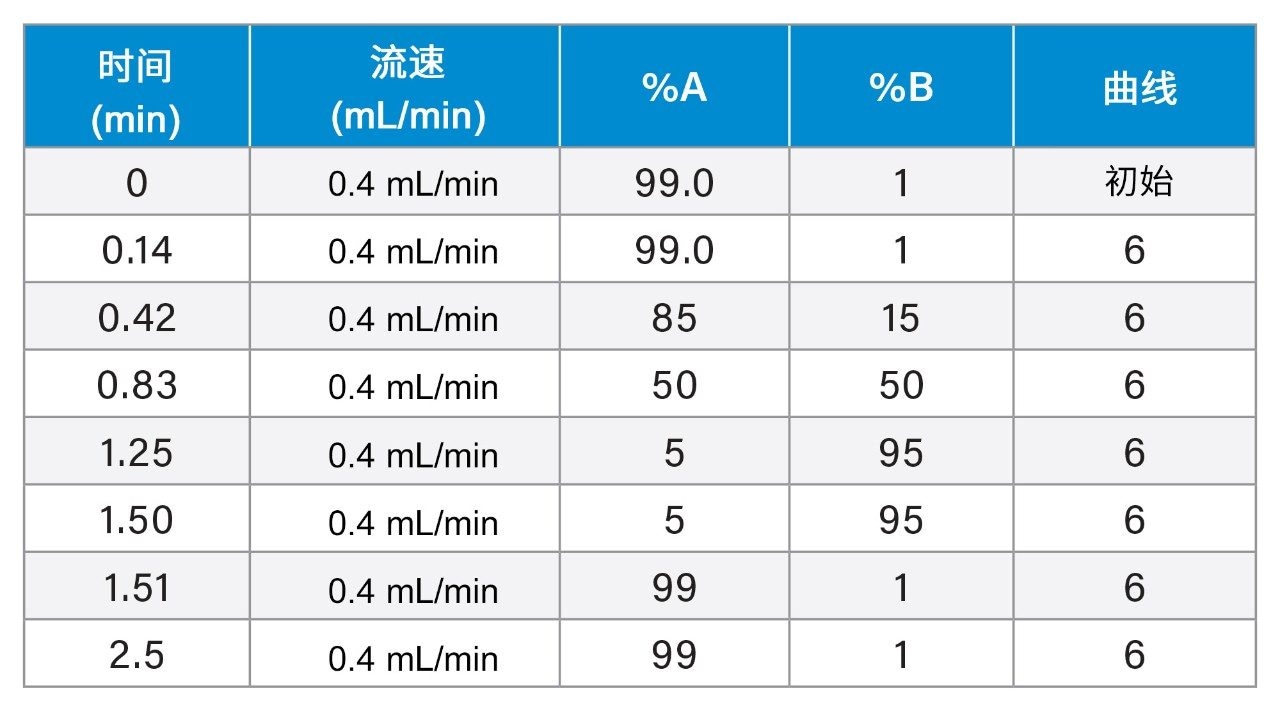
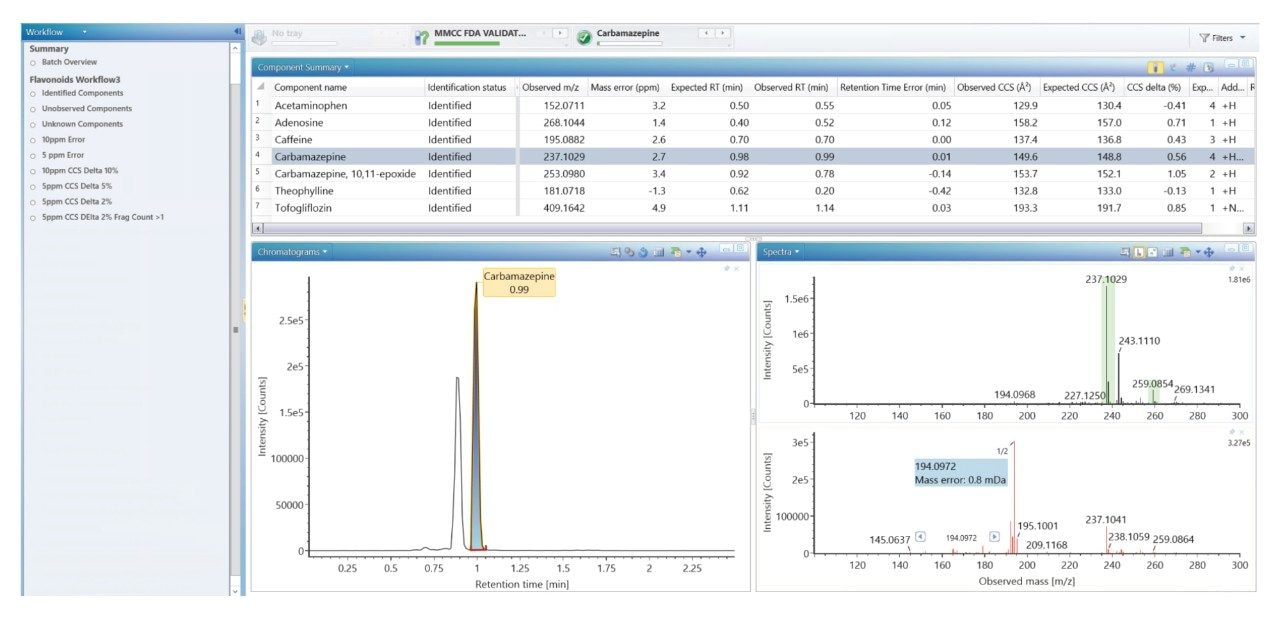
参考之前描述的质谱数据库生成策略2,利用快速梯度微孔超高效液相色谱-离子淌度质谱联用系统(RGM-UPLC-IM-MS)表征一组FDA批准的小分子药物。生成的数据库对串联质谱、ToF或IM分析均有帮助。采用的策略为所表征的分析物提供了保留时间(tr)、母离子、子离子和碰撞截面(CCS)数据。之前使用常规UPLC方法生成质谱数据库,总运行周期为12 min,使用2.1 mm × 100 mm色谱柱,应用的洗脱液流速为0.5 mL/min。本研究使用快速微孔代谢谱分析UPLC-MS方法生成了等效的快速梯度微孔UPLC-IM-MS数据库,该方法采用1 mm × 50 mm色谱柱和0.4 mL/min的洗脱液流速,这些条件使总分析周期缩短至2.5 min。常规和快速梯度UPLC-MS所提供的峰容量分别为150和503。
高分辨率质谱仪(HRMS),例如四极杆飞行时间(Q-Tof)质量分析器,已成为临床、法医毒理学和代谢物鉴定研究领域中越来越常用的筛查工具4,5 。在非靶向“全扫描”数据采集中,只需一次分析即可完成数千次检测,随后进行回顾性靶向数据分析6。对于许多研究领域(例如农药、真菌毒素和天然植物毒素的检测)而言,提高样品通量、时间效率和降低成本势在必行,由此推动了多类别化合物分析的发展7。鉴于样品复杂性是一项常见的挑战,这样的目标也存在于代谢表型分析中,该分析的目的是在分子水平了解基因型、环境与生活方式之间的相互作用。这些研究可能包括数以千计的临床前代谢/毒理学、临床和流行病学样品3。
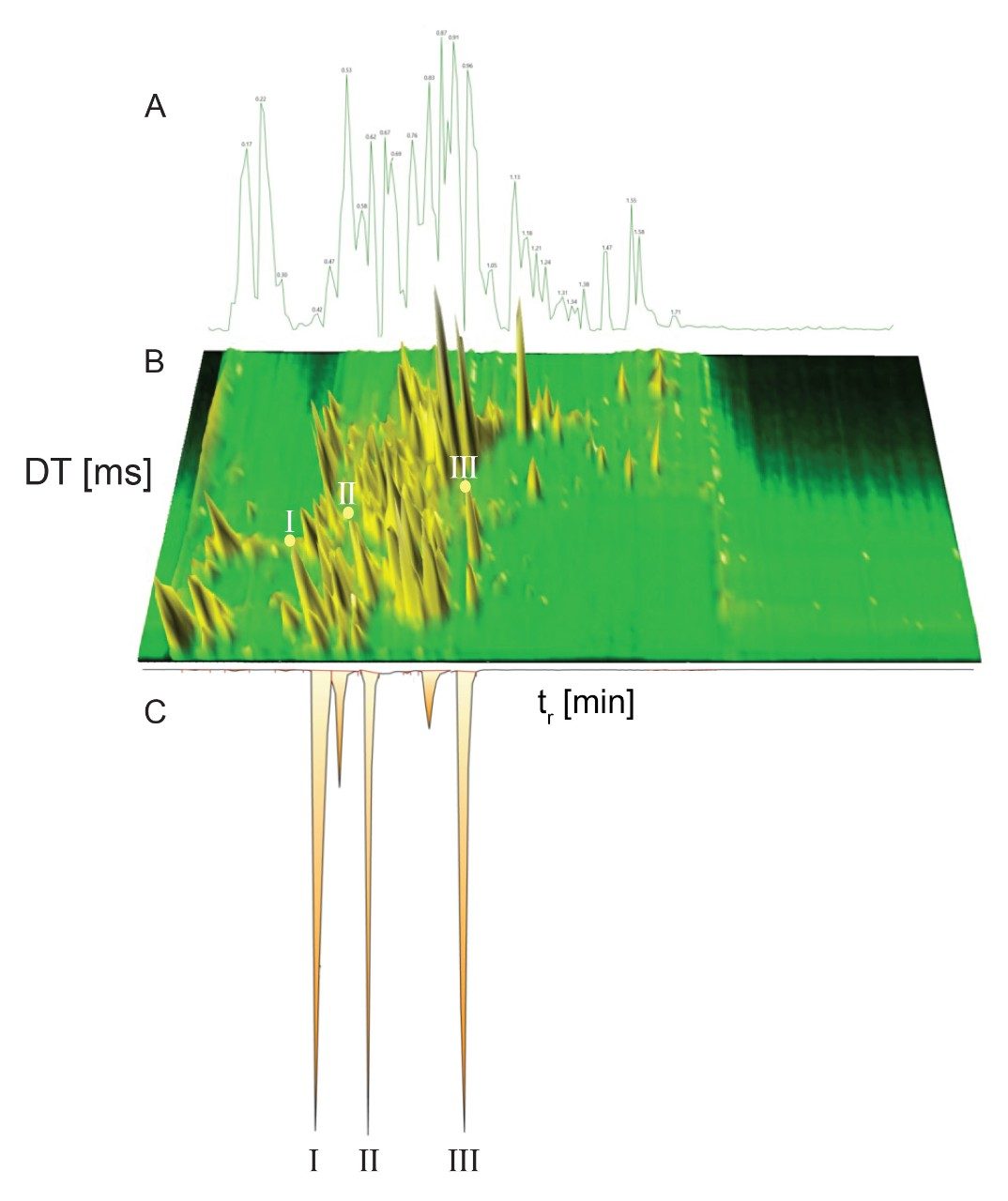
Dwivedi等人探讨了LC、MS与IM的正交性,表明峰容量提高至2~10倍(具体取决于MS和IM分辨率)8。将RGM-UPLC方法与离子淌度质谱法相结合,可以增强峰容量、特异性和分析灵活性,样品通量提高至5倍,同时保持该策略的时间效率9,10。UPLC-IM包括离子淌度技术(MS分析前的气相分离)与UPLC技术(中性物质分离)11,12。UPLC(秒)、IMS(毫秒)和飞行时间MS(微秒)的时间尺度符合复杂样品的高通量分析要求。化合物的离子淌度分离是气相离子在质谱仪中充满气体的行波离子淌度(TWIM) RF离子导向装置内发生分离,再进入质量分析器。淌度分离通过使用相对较弱的电场驱动离子包穿过惰性缓冲气体(氮气)来实现。离子与缓冲气体之间的碰撞次数导致漂移时间差异。分离结果基于沿RF离子导向装置施加的重复DC脉冲;离子周期性地被脉冲或波超越,淌度较低的物质比淌度较高的物质更频繁地被超越。因此,穿越装置的时间取决于淌度,并受离子质量、电荷和形状等因素的控制。离子淌度在CCS(碰撞截面,一种补充性的鉴定指标)之外,还为LC(疏水性)和MS (m/z)提供了额外的分离维度。CCS在提高鉴定特异性方面的作用已在各种应用中得到证实,包括代谢物鉴定、农药分析、真菌毒素筛查、药用植物物种形成研究和食品添加剂分析10,13-17。
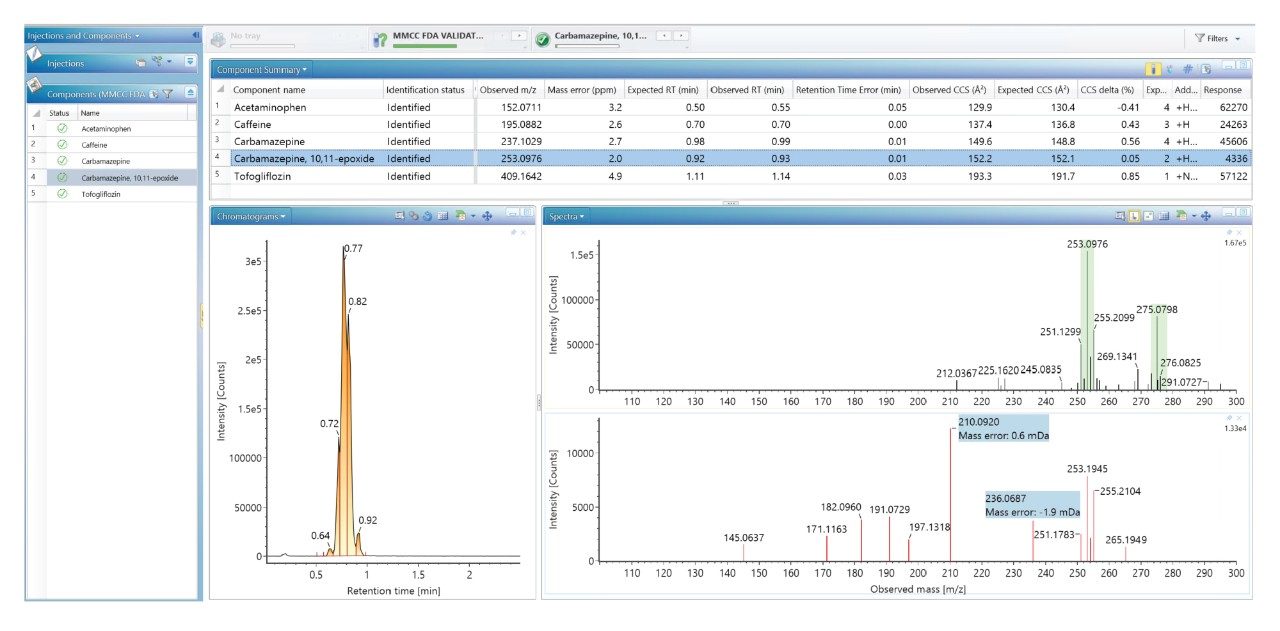
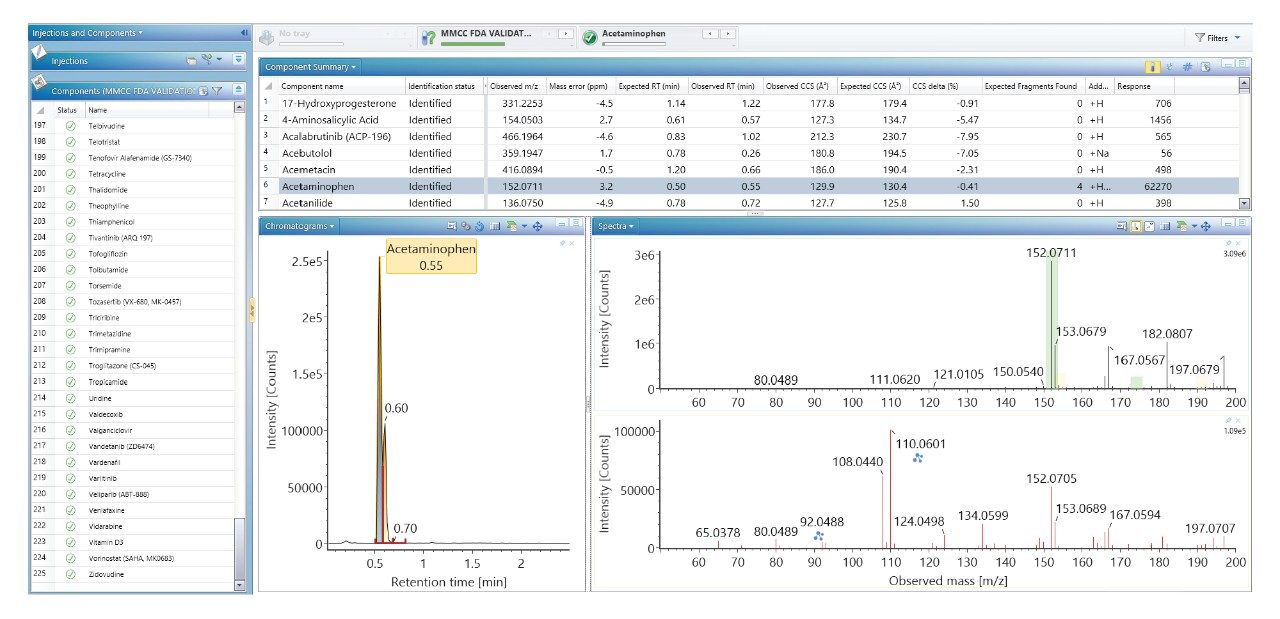
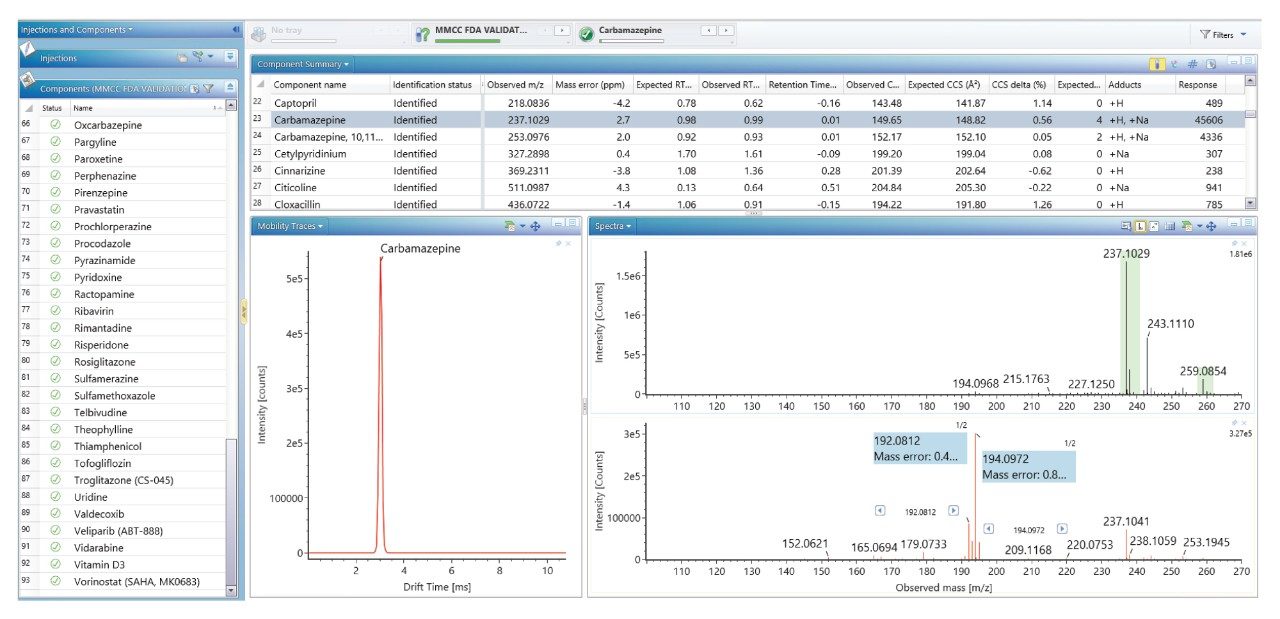
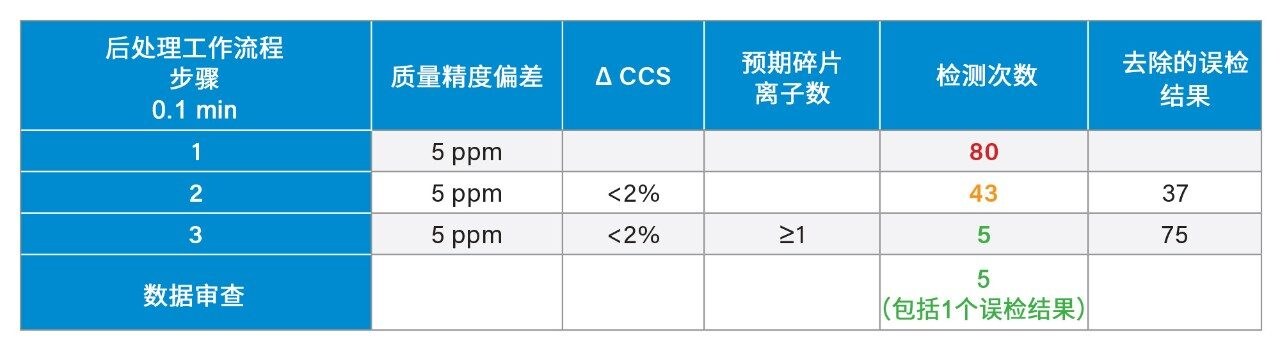
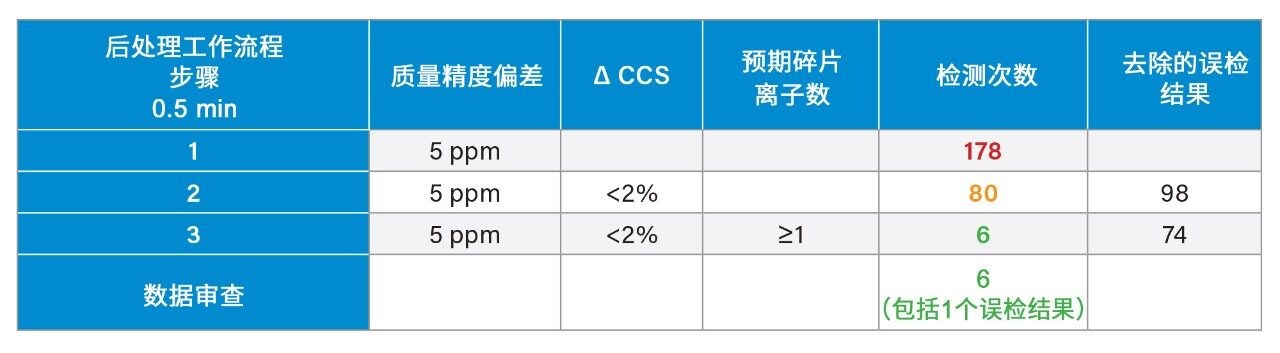
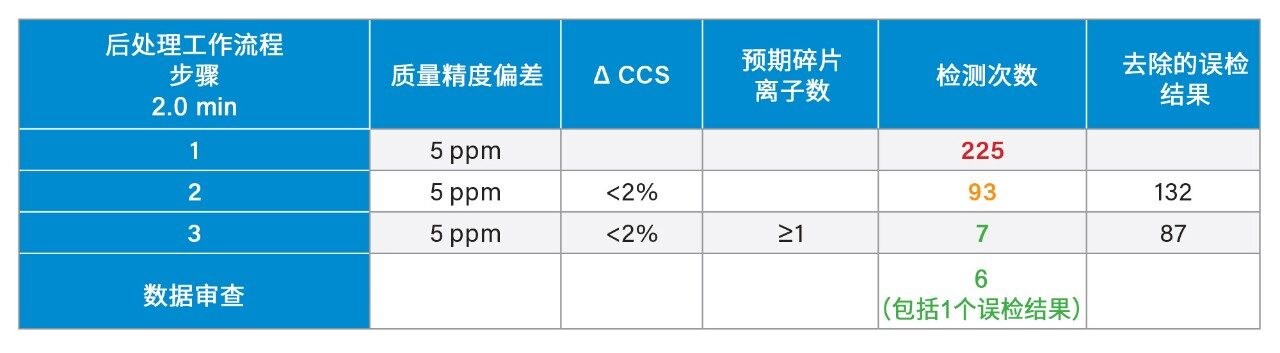
我们使用一组FDA批准的小分子药物数据库对人类健康志愿者的尿样进行药物筛查,以鉴定施用的药物化合物并将其与复杂生物基质中的内源性化合物区分开来。通过比较常规UPLC-IM-MS和RGM-UPLC-IM-MS数据库的应用,评估了该方法的可行性。