ケーススタディ:Arc HPLC システムを用いた USP 有機不純物分析法のグローバルラボ間試験における予期せぬ結果の調査
要約
グローバル経済を生き抜く多くのラボでは、分析法を世界中で移管する必要があります。分析法移管には、装置の可用性、サンプル前処理、移動相調製など、分析法に関連する変動要因を管理するための調整されたアプローチと戦略が必要です。ただし、いかに優れた戦略を取っても、特に地理的な違いや環境、言語の違いを考慮すると、世界中のラボ間での変動を管理することは困難な場合があります。外れ値や予期せぬ結果が発生した場合、分析法に影響を及ぼす可能性のある要因を十分に理解することが、その調査と解決に重要です。このアプリケーションノートでは、USP 有機不純物分析法のグローバルなラボ間分析法移管を、世界中の 8 つの施設で実施しました。管理戦略と分析法の性能に関する重要な情報を元の施設から入手し、試験に組み入れました。管理戦略のさらに詳細な分析は、アプリケーションノート『リスクベースのアプローチを用いた、USP 有機不純物分析法の Arc HPLC へのグローバルラボ間分析法移管』に記載しています1。 管理戦略を整備することで、システム適合性要件がすべてのラボで日常的に満たされていました。一方、分析法とシステム変動要因を理解することで、予期せぬ結果の調査と原因についての指針が得られました。
アプリケーションのメリット
- Arc HPLC システムが、システム間で簡単に再現できる高品質のデータ性能を提供
- XBridge C8 カラムの採用により、USP クエチアピン不純物分析法をシステム間で移管する際のカラム間の変動が低減
- Empower 3 ソフトウェアのトレーサビリティ機能により、試験に関与した各ラボが指示の概要に適切に準拠したという記録を提供
はじめに
グローバル経済を生き抜く多くのラボでは、分析法を世界中で移管する必要があります。分析法移管には、装置の可用性、サンプル前処理、移動相調製など、分析法に関連する変動要因を管理するための調整されたアプローチが必要です。ただし、いかに優れた戦略を取っても、特に地理的な違いや環境、言語の違いを考慮すると、世界中のラボ間での変動を管理することは困難な場合があります。外れ値や予期せぬ結果が発生した場合、分析法に影響を及ぼす可能性のある要因を十分に理解することが、その調査と解決に重要です。
このアプリケーションノートでは、USP 有機不純物分析法のグローバルなラボ間分析法移管を、世界中の 8 つの施設で実施しました。管理戦略と分析法の性能に関する重要な情報を元の施設から入手し、試験に組み入れました。管理戦略を整備することで、システム適合性要件がすべてのラボで日常的に満たされていました。一方、分析法とシステム変動要因を理解することで、予期せぬ結果の調査と原因についての指針が得られました。これらの予期せぬ結果について、このアプリケーションノートで詳しく説明します。
実験方法
この分析法は、クエチアピンフマル酸塩不純物の USP モノグラフ2 に基づいており、調整は行いませんでした。
サンプルの説明
この分析法には、システム適合性レファレンス標準試料(RS)およびクエチアピンフマル酸塩の RS の両方が必要です。システム適合性溶液は、USP クエチアピンシステム適合性 RS(USP 製品番号:1592715)から調製し、クエチアピン、クエチアピンデスエトキシ(1 ~ 5%)、類縁物質 G、および類縁物質 B の混合物で構成されています。システム適合性溶液は、希釈液(86:14 移動相 A/移動相 B)中に 1 mg/mL の濃度で調製しました。
標準溶液は、USP クエチアピンフマル酸塩 RS を使用して、希釈液中に 0.001 mg/mL の濃度になるように調製しました。
原薬は、Hangzhou Think Chemical Co., Ltd. から入手し、有効期限を過ぎていました。サンプルは、移動相 A 中に 1.0 mg/mL の濃度で調製しました。
分析条件
|
カラム: |
XBridge 8、3.5 µm、4.6 mm × 150 mm(ウォーターズ製品番号:186003055) |
|
|
カラム温度: |
45 ℃ |
|
|
サンプル温度: |
10 ℃ |
|
|
注入量: |
20 μL |
|
|
検出: |
250 nm |
|
|
データレート: |
10 Hz |
|
|
流速: |
1.5 mL/分 |
|
|
分析時間: |
70 分 |
|
|
バッファー: |
3.1 g/L 酢酸アンモニウム水溶液。溶液各 1 L に 2 mL の 25% 水酸化アンモニウムを添加。pH は 9.2 以上 |
|
|
移動相 A: |
25:75 ACN:バッファー |
|
|
移動相 B: |
アセトニトリル |
|
|
ニードル洗浄溶媒: |
50:50 水:アセトニトリル |
|
|
パージ溶媒: |
50:50 水:アセトニトリル |
|
|
シール洗浄溶媒: |
90:10 水:アセトニトリル |
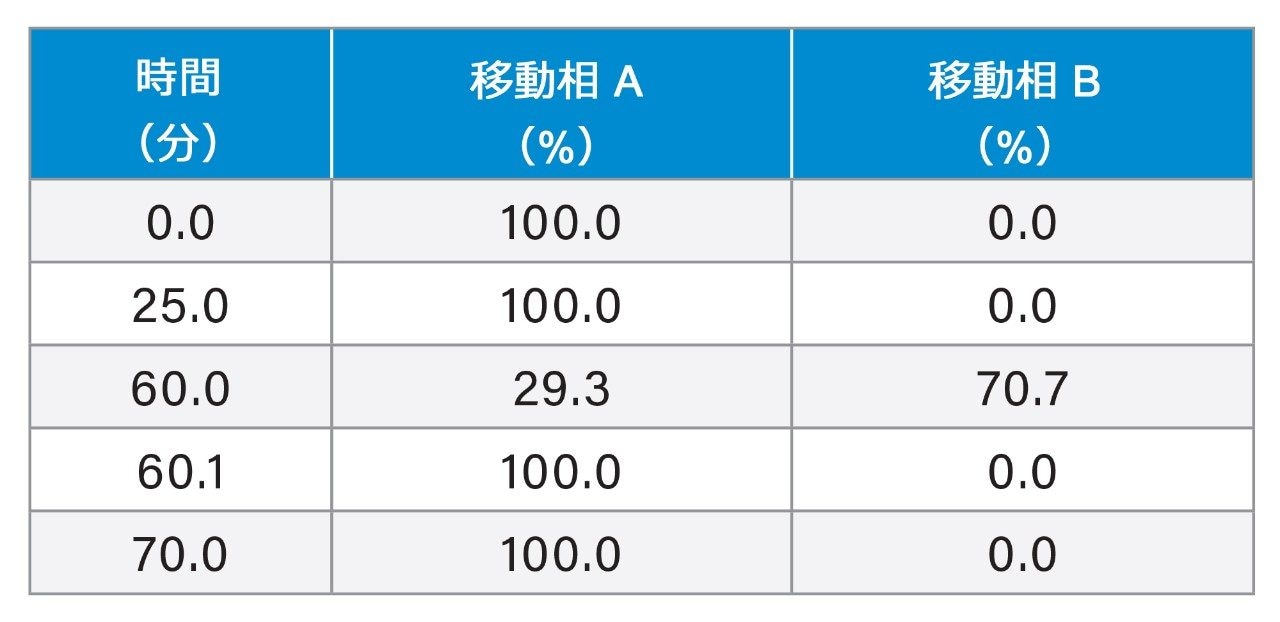
Gradient
データ管理
Empower 3 クロマトグラフィーデータソフトウェア
システム設定
|
システム: |
Arc HPLC システム(QSM-R、FTN-R、CHC) |
|
|
検出: |
2998(PDA)または 2489(TUV) |
|
|
設定: |
パッシブプレヒーター(推奨) |
|
|
フローセル: |
分析 |
結果および考察
試験
USP クエチアピンフマル酸塩不純物分析法の分析法移管2 を、世界中の施設にわたって実施しました。移管元ラボは米国マサチューセッツ州 Milford にあり、7 つの移管先ラボはマサチューセッツ州 Milford の第 2 ラボ、トルコ、インド、シンガポール、中国、フランス、米国ノースカロライナ州にありました(表 1)。この試験には、システム適合性要件の確認および原薬の定量分析が含まれました。すべての分析を、可変波長(TUV)2489 検出器またはフォトダイオードアレイ(PDA)2998 検出器を搭載した Arc HPLC システムで実行しました。各分析は、モノグラフに記載されているように、システム適合性要件を用いて評価するとともに、原薬を分析し、不純物の分析を比較して評価しました。システム適合性基準は、システム適合性溶液の 2 つのクリティカルペアの分離、標準溶液のピークのテーリング、保持時間の %RSD、面積の %RSD に基づきました。
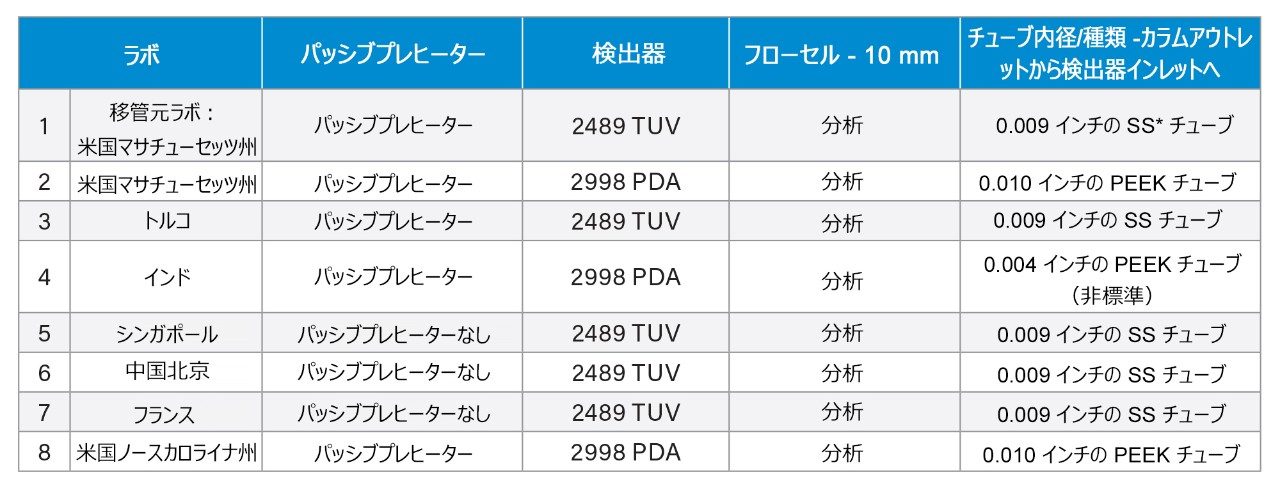 表 1. 各ラボの所在地およびシステム構成
表 1. 各ラボの所在地およびシステム構成
この試験では、世界中で分析法の再現性を確保するための管理戦略を設定しました。例えば、すべての関与するラボには、標準試料、カラム、サンプル製剤を含む同一の試薬キットが送付されました。(ただし、すべてのカラムが同じロットとは限りませんでした。)標準的な操作手順プロトコルを作成し、多くのラボに配布しました。元の USP 分析法を維持するために、移動相のろ過や、分析法に関連する化学物質(水酸化アンモニウムや酢酸アンモニウムなど)は未開封の容器または最近開封した容器を使用することなど、移動相の調製について慎重に記載しました。これらの管理戦略およびその実施方法の詳細については、アプリケーションノート『リスクベースのアプローチを用いた、USP 有機不純物分析法の Arc HPLC へのグローバルラボ間分析法移管』を参照してください2。
クエチアピン不純物分析法は、移管元ラボでベースライン化され、他のラボに移管されました。各ラボは、システム適合性が満たされていることを確認するためにデータを解析しましたが、すべての定量的データは、移管元ラボの施設で単一の解析メソッドを使用して解析されました。すべてのデータをレビューしたところ、不一致や予期せぬ結果があり、さらに調査する必要がありました。
ケーススタディ 1:保持時間の違い
問題
システム適合性溶液により、クエチアピン(API)とクエチアピンデスエトキシというクリティカルペアの分離、および類縁物質 G と類縁物質 B の分離を評価します。この試験の 8 つのラボは、図 1 のクロマトグラムで分かるように、すべてこのクリティカルペアの要件を満たしていました。クロマトグラムを分析すると、すべてのラボの結果で保持時間が 1% 以内と再現性がありました。ただし、ラボ 7 では、他のすべてのラボと比較して、クロマトグラフィーにおける保持時間が大きくシフトしていました。ラボ 7 ではシステム適合性要件を満たしていましたが、保持時間のシフトに対して考えられる原因を特定するために調査が行われました。
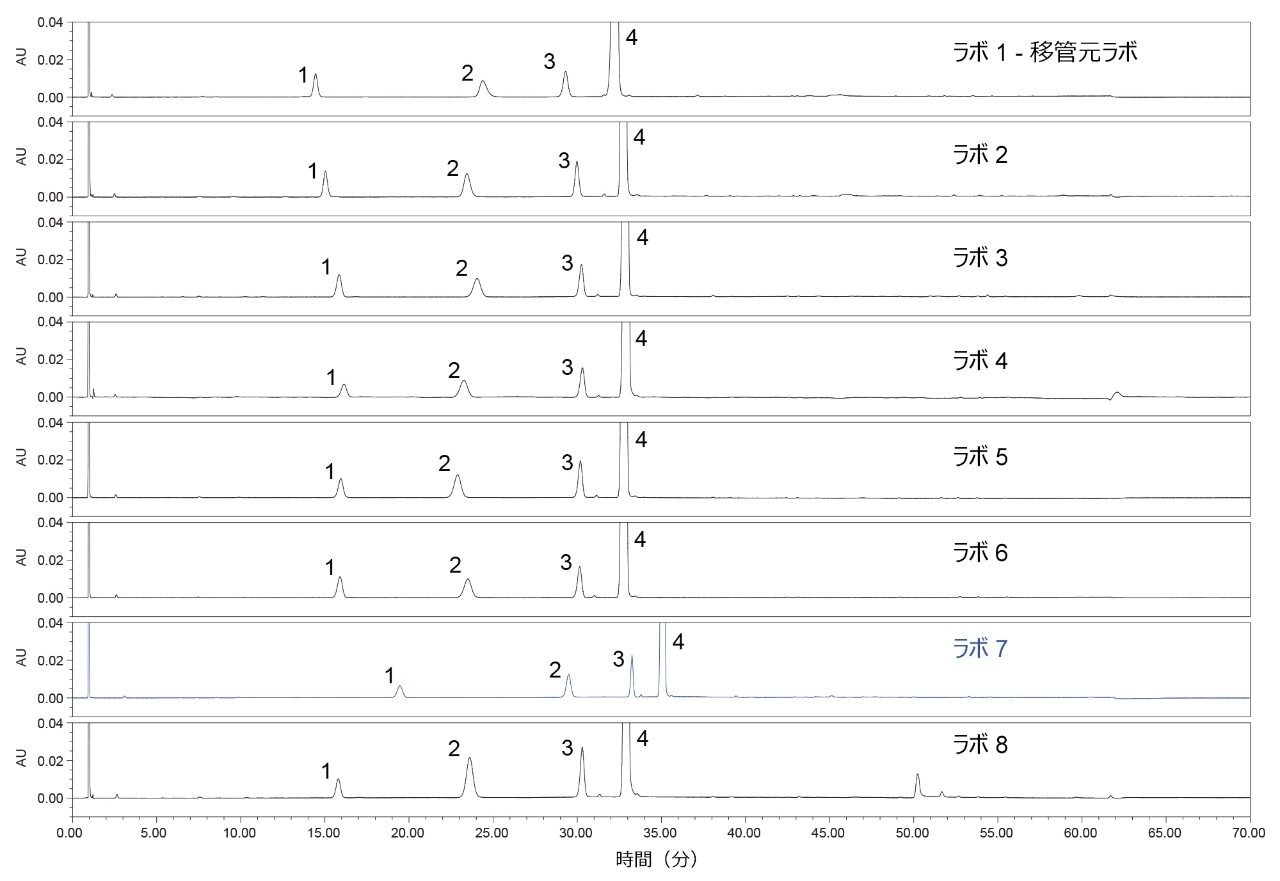 図 1. この試験に参加した 8 つのラボで得られたシステム適合性溶液の分析結果。ピーク 1:類縁物質 G、ピーク 2:類縁物質 B、ピーク 3:クエチアピンデスエトキシ、ピーク 4:クエチアピン(API)。
図 1. この試験に参加した 8 つのラボで得られたシステム適合性溶液の分析結果。ピーク 1:類縁物質 G、ピーク 2:類縁物質 B、ピーク 3:クエチアピンデスエトキシ、ピーク 4:クエチアピン(API)。
調査
- 装置
すべてのラボからのクエチアピンピークの保持時間をプロットすると(図 2)、ラボ 7 の保持時間のシフトが 2 分を超えていることが分かりました。保持時間シフトの原因を特定するために、最初のステップとして、各システムからのシステム圧力トレースを調べました(図 3)。ラボ 7 のシステム圧力が他のラボよりも高かった場合は、圧力の上昇が保持時間に影響を及ぼした可能性があります。ラボ 7 のシステム圧力は他のラボと同等であったため、保持時間のシフトを引き起こしたのはシステム圧力ではありませんでした。
ただし、システム圧力を調査したところ、ラボ 4 の Arc HPLC システムは、他のすべてのシステムおよびラボよりも背圧が大幅に高く、システム圧力が約 20% 高いことが分かりました。システムを調査したところ、システム構成と相関していることが分かりました。他のすべてのラボでは、カラムアウトレットから検出器までに 0.009 インチまたは 0.010 インチの標準構成チューブが使用されていましたが、ラボ 4 のシステムでは、カラムアウトレットと検出器の間が内径 0.004 インチの非標準チューブ(表 1 を参照)で構成されました。小さいチューブ内径の影響は、分離能の低下と背圧の上昇の両方に表れています。したがって、システムの圧力とピークの保持時間の間に相関関係はありません。ただし、このアプリケーションノートを通して分かるように、すべてのシステム適合性要件は同等でした。
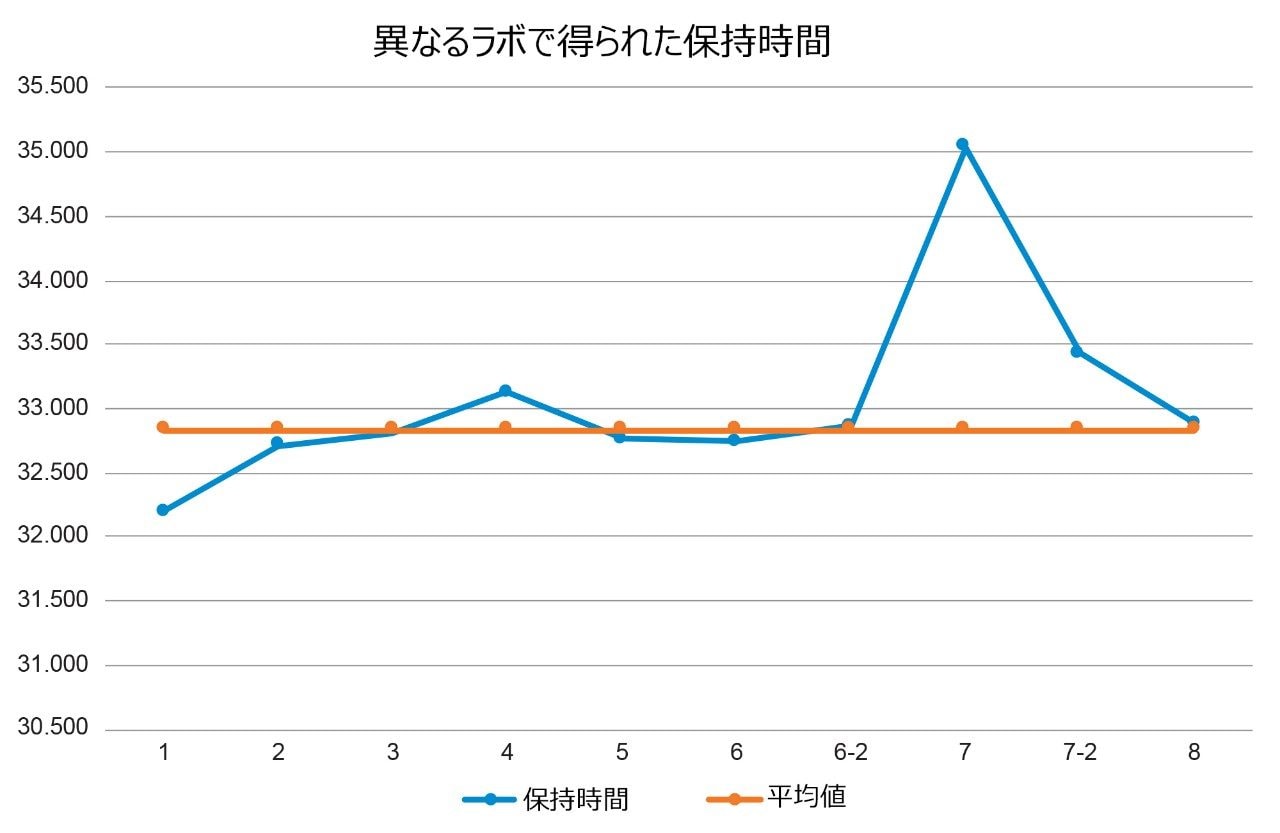 図 2. 8 つの参加ラボすべてにおける標準溶液でのクエチアピンピークの保持時間。ラボ 7 での最初の保持時間シフトは、他のラボより 2 分超長くなっています。
図 2. 8 つの参加ラボすべてにおける標準溶液でのクエチアピンピークの保持時間。ラボ 7 での最初の保持時間シフトは、他のラボより 2 分超長くなっています。
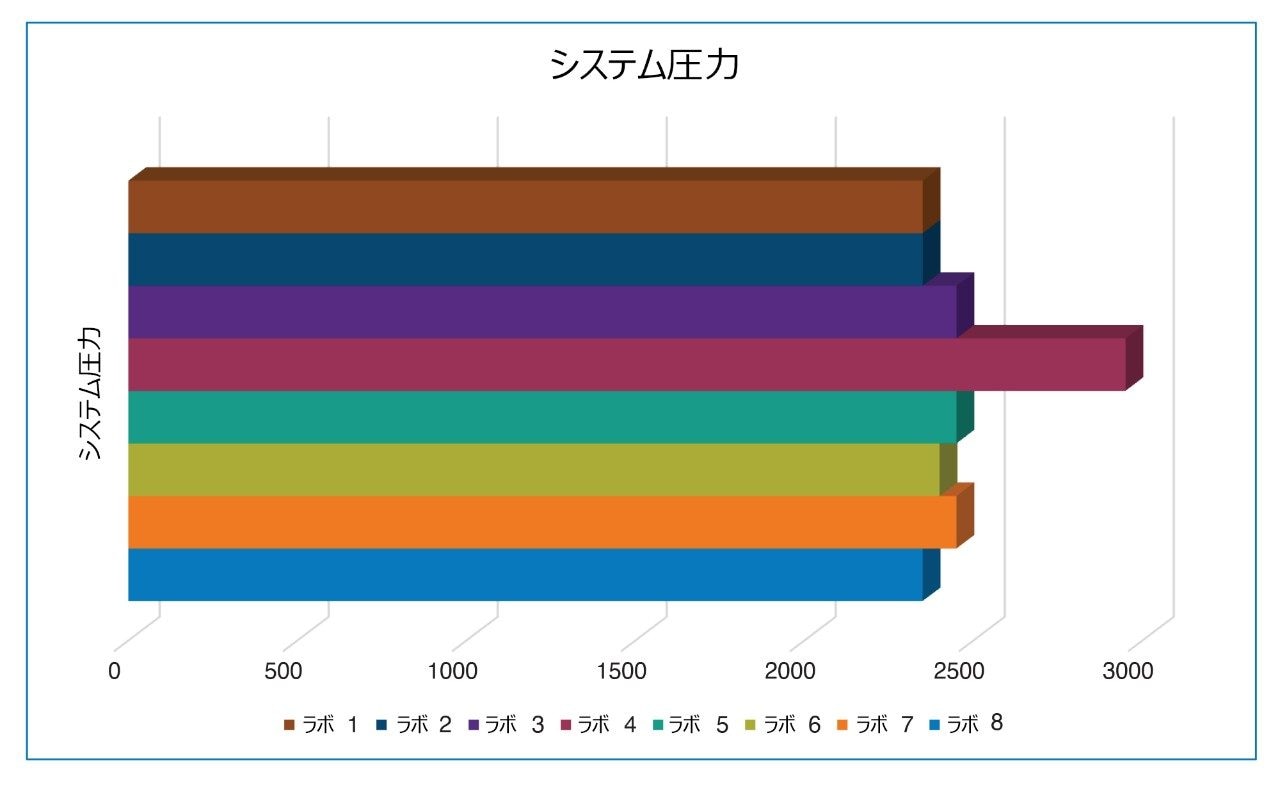 図 3. 8 つの参加ラボすべてにおける Arc HPLC システムでのシステム圧力
図 3. 8 つの参加ラボすべてにおける Arc HPLC システムでのシステム圧力
- 手順/移動相の調製
保持時間シフトについて、考えられる原因としてシステム圧力が除外されたことにより、根本原因を特定するためにより多くの情報が必要になったため、ラボ 7 では分析を繰り返し行いました(図 4)。2 回目の分析では、ラボ 7-2 に関する上記の図 2 に示すように、ピークの保持が他のラボにはるかに近づいていることが示されました。このことは、移動相が保持時間シフトの原因である可能性を示しています。また、ラボ 7 にはキャリブレーション済みの pH メーターがないことが分かり、バッファーの正確な pH は不明でした。最初の分析では、ラボ 7 は SOP の指示に従って 4 mL の水酸化アンモニウムを添加していましたが、2 回目の分析では、pH が 9.2 という目的の値ではないと考え、5 mL の水酸化アンモニウムをバッファーに添加しました。したがって、バッファーの pH が、保持時間のシフトの原因の 1 つである可能性があります。もう 1 つの可能性のある原因として、この論文には示していない以前の試験に基づき、移動相 A の有機溶媒とバッファーの比率である可能性があります。逆相分析法について予想されるように、アセトニトリル含量を相対的に増加させるとピークの溶出時間がより早い方にシフトし、アセトニトリルを相対的に減少させるとピークの保持時間がより遅い方にシフトします。
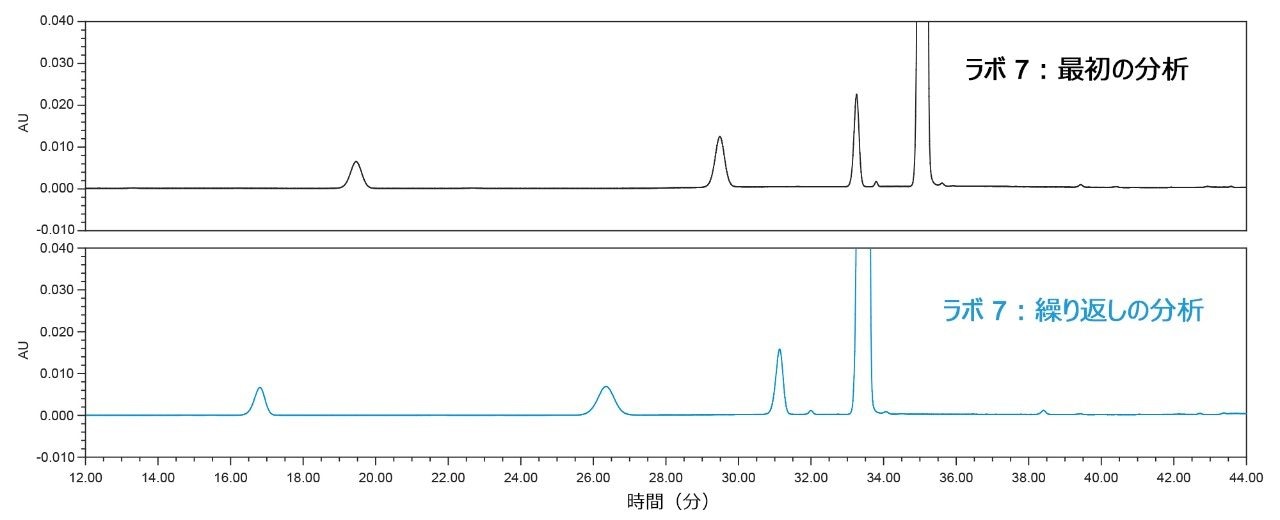 図 4. ラボ 7 での最初の分析(上段)およびラボ 7 での保持時間が右にシフトした 2 回目の分析(下段)で得られたシステム適合性溶液のクロマトグラム
図 4. ラボ 7 での最初の分析(上段)およびラボ 7 での保持時間が右にシフトした 2 回目の分析(下段)で得られたシステム適合性溶液のクロマトグラム
pH が保持時間に影響を及ぼしているかどうかを判定するため、移管元のラボでは、ピークの保持時間に対する pH の影響を判定するための試験を実施しました。移管元のラボでこの分析法を定期的に行う際に、要求されている 4 mL の水酸化アンモニウムを添加した後のバッファーの pH は 9.2 です。移管元のラボで 5 mL の水酸化アンモニウムをバッファーに添加したところ、pH の測定値は 9.4 になりました。追加のバッファーを pH 9.8 で調製しましたが、これにはさらに約 4 mL の水酸化アンモニウムを追加する必要がありました。異なる pH のバッファー溶液をそれぞれ調製した後、モノグラフの記載に従って移動相 A を調製し、保持時間に対する pH 9.2、pH 9.4、pH 9.8 の影響を評価するために分析を行いました。pH 値が異なるシステム適合性溶液で得られたクロマトグラムを図 5 に示します。図 5 に示すように、pH は確かにピークの保持時間に影響していますが、その影響はラボ 7 で最初に見られた保持時間のシフトほど大きくはありません。この情報および移動相 A の有機組成を調べた以前の試験に基づいて、有機物とバッファーの比率が保持時間シフトの原因である可能性が最も高いという仮説が導かれます。
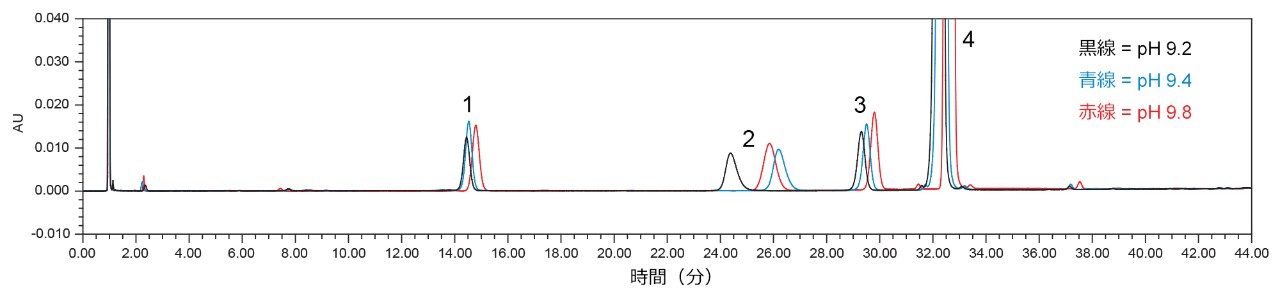 図 5. システム適合性溶液での移動相 pH の影響の評価結果。バッファーは pH 9.2(黒)、pH 9.4(青)、および pH 9.8(赤)。ピーク 1:類縁物質 G、ピーク 2:類縁物質 B、ピーク 3:クエチアピンデスエトキシ、ピーク 4:クエチアピン(API)。
図 5. システム適合性溶液での移動相 pH の影響の評価結果。バッファーは pH 9.2(黒)、pH 9.4(青)、および pH 9.8(赤)。ピーク 1:類縁物質 G、ピーク 2:類縁物質 B、ピーク 3:クエチアピンデスエトキシ、ピーク 4:クエチアピン(API)。
- 解決策
移動相の調製による変動を低減するには、クエチアピン不純物分析法用に規定された器具で移動相溶液を正確かつ精密に測定することが重要です。分析者間のばらつきを完全に排除することは不可能ですが、移動相を調製する器具の正確なサイズとグレードを指定することにより、ばらつきの一部を低減することができます。溶液ごとの調製のばらつきを最小限に抑えることで、再現性が向上します。
ケーススタディ 2:面積の %RSD の違い
問題
標準溶液を使用して、テーリング係数、保持時間の %RSD、および面積の %RSD のシステム適合性要件を評価します。アプリケーションノート『リスクベースのアプローチを用いた、USP 有機不純物分析法の Arc HPLC へのグローバルラボ間分析法移管』1に記載されているように、システム適合性の結果(表 2)は、システム適合性要件の範囲内でした。ただし、結果をレビューしたところ、3 つのデータポイントは外れ値であるように思われました。具体的には、ラボ 2、ラボ 4、ラボ 8 で面積の RSD の割合が高くなっています。これらの値は要件を満たしていましたが、根本原因をさらに理解するために調査を行いました。
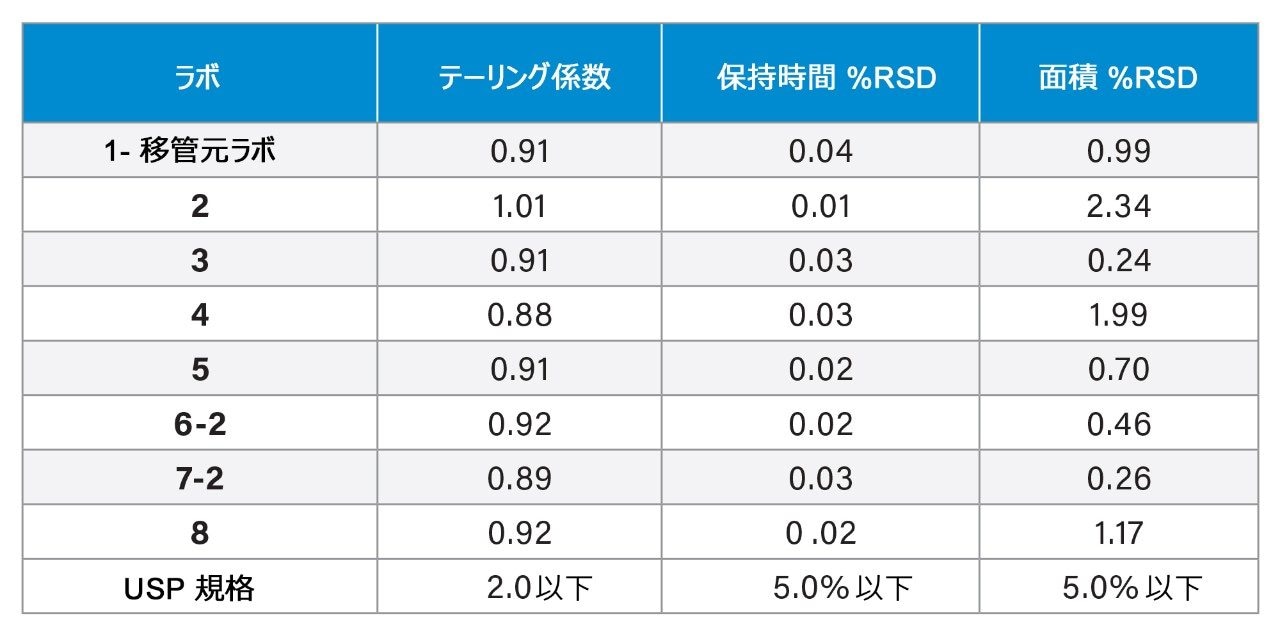 表 2. 8 つのラボすべての標準溶液から得られたシステム適合性の結果
表 2. 8 つのラボすべての標準溶液から得られたシステム適合性の結果
調査
ラボから得られたシステム構成の情報によると、%RSD が大きい 3 つのラボで PDA 検出器が使用されていることが分かりました。3 台の PDA 検出器で得られたクロマトグラムを TUV 検出器の結果と比較したところ、PDA データには TUV データよりも乱れが多く見られました(図 6)。TUV 検出器は、PDA よりも感度が高く、直線性の範囲が広いことから、TUV のベースラインの低いノイズは、予想通り均一性が高くなります。PDA で見られるベースラインノイズが大きいほど、標準溶液(1μg/mL)の面積の変動が大きくなります。
この仮説を検証するため、クエチアピン不純物分析法を 2 台の個別の検出器を備えた単一の Arc HPLC システムで試験しました。まず、TUV(2489)検出器を備えた Arc HPLC で分析を行い、次に検出器を PDA(2998)に交換しました。結果を比較すると(図 7)、単一の Arc HPLC システムで、PDA 検出器は TUV 検出器よりも大きなノイズを示しました。また、表 3 の結果では、他のテーリング係数と保持時間の %RSD の結果は同等であった一方、面積 %RSD に有意差があることを示しています。 データは、ベースラインノイズの違いが検出器によるものであり、サンプル、移動相、またはシステムの変動によるものではないことを示しています。この分析では TUV の方が良好な性能を示しましたが、いずれの検出器もシステム適合性要件の仕様を満たしていました。
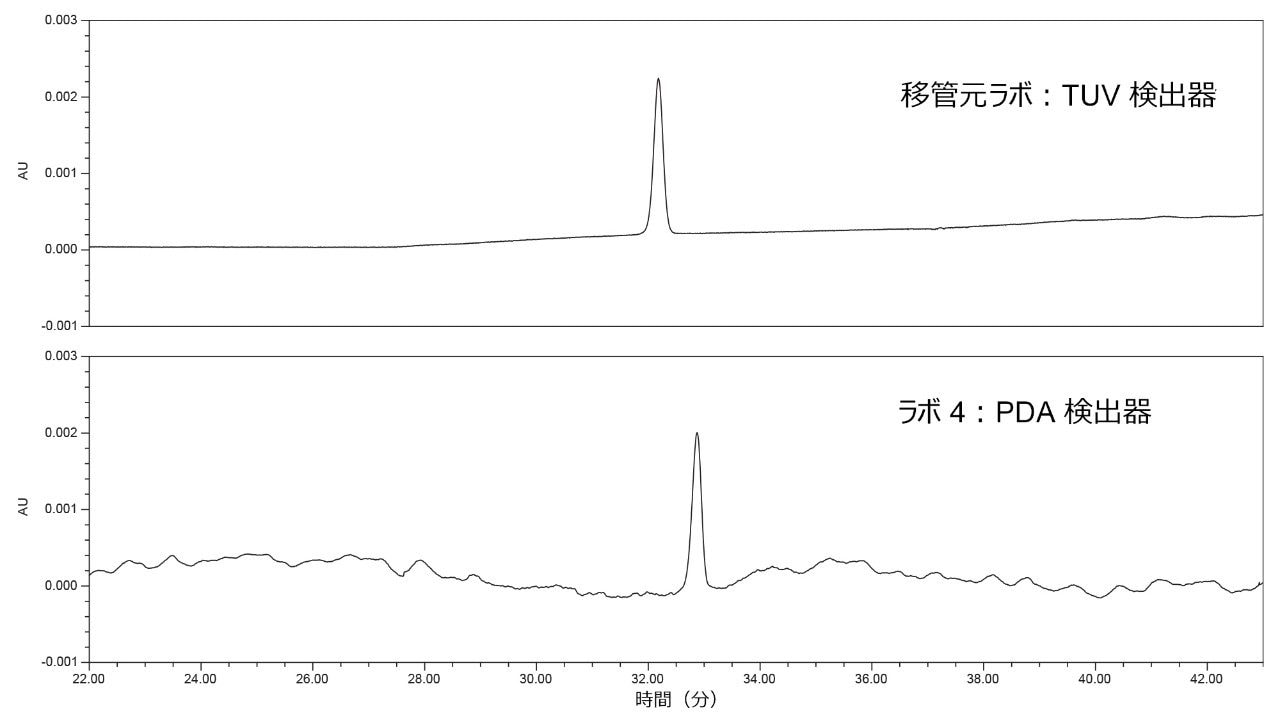 図 6. TUV 検出器を使用している移管元ラボであるラボ 1(上段)および PDA 検出器を使用しているラボ 4(下段)で得られた標準溶液のクロマトグラム
図 6. TUV 検出器を使用している移管元ラボであるラボ 1(上段)および PDA 検出器を使用しているラボ 4(下段)で得られた標準溶液のクロマトグラム
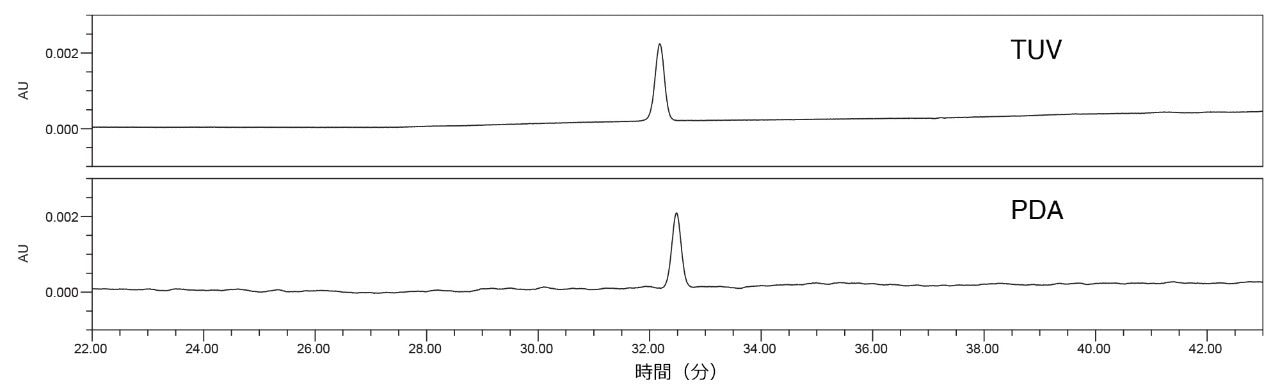 図 7. 移管元ラボ、ラボ 1 で TUV 検出器を搭載した Arc HPLC を使用し(上段)、および PDA 検出器を搭載した同じ Arc HPLC を使用して(下段)得られた標準溶液のクロマトグラム
図 7. 移管元ラボ、ラボ 1 で TUV 検出器を搭載した Arc HPLC を使用し(上段)、および PDA 検出器を搭載した同じ Arc HPLC を使用して(下段)得られた標準溶液のクロマトグラム
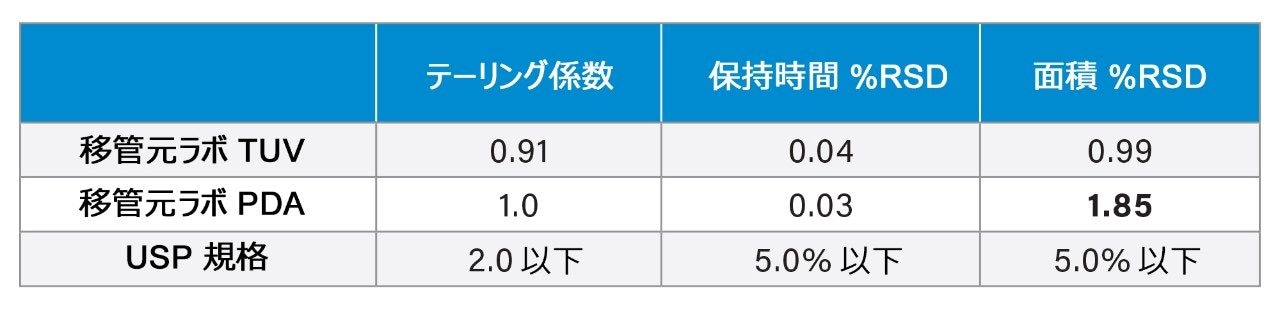 表 3. TUV 検出器と PDA 検出器を搭載した単一の Arc HPLC システムで得られた標準溶液の分析結果
表 3. TUV 検出器と PDA 検出器を搭載した単一の Arc HPLC システムで得られた標準溶液の分析結果
解決策
将来の試験のために考えられる解決策またはアプローチとして、TUV と PDA の両方で低濃度のサンプルを試験し、両方の検出器で感度または精度の要件を満たすよう確認することが挙げられます。この例において、通常のトラブルシューティング実験では、面積の大きい %RSD は通常、インジェクターまたはその他のコンポーネントに起因すると考えられますが、システム構成の調査をまず行うことにより、一定の傾向が認められました。この例では、クロマトグラフィーの結果とは無関係に、システム全体の調査が必要であることを示しています。また、この結果では、特定の検出器が必要とされない場合は、PDA と TUV の両方で分析法を評価する必要があることも示しています。これにより、今回の場合のように、両方の検出器で分析法のシステム適合性要件を満たすことができます。
ケーススタディ 3:定量結果の違い
問題
サンプル溶液または原薬を分析して、不純物のクエチアピンデスエトキシおよび未知不純物の定量結果を得ました。1 つを除くすべてのラボで、同等の定量結果が得られました(表 4)。この例では、データをレビューしたところ、2 つの不純物(クエチアピンデスエトキシと未知不純物)の割合が、ラボ 6 でははるかに低い値となりました。これらの結果により、不純物の値が低くなる原因を特定するためにデータを詳しく調べました。
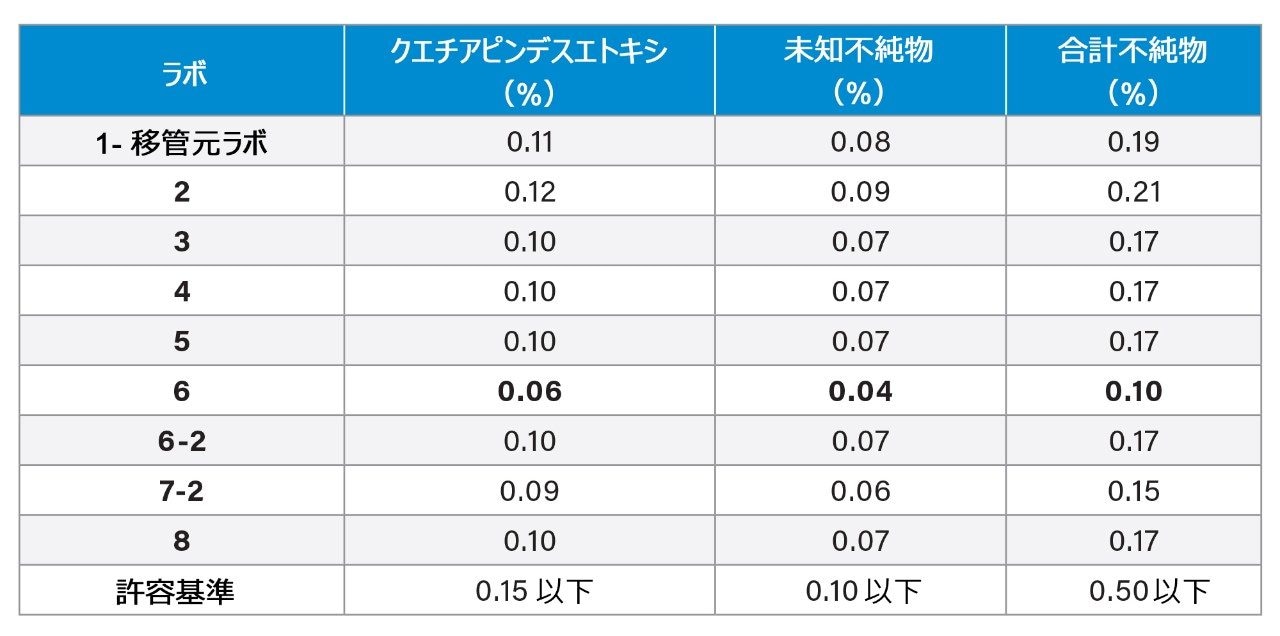 表 4. 8 つの参加ラボすべてで得られたサンプル溶液の、クエチアピンデスエトキシおよび未知不純物のピークについての定量結果
表 4. 8 つの参加ラボすべてで得られたサンプル溶液の、クエチアピンデスエトキシおよび未知不純物のピークについての定量結果
調査
最初に行ったステップでは、すべてのラボで得られた標準クロマトグラムを比較し(図 8)、各ラボで得られた標準ピーク面積を比較しました(図 9)。サンプルの不純物濃度は、標準溶液に対する標準面積の割合として計算されるため、標準クロマトグラムを調査しました。ラボ 6 の標準ピーク面積は、他のすべてのラボの結果よりも大きいことが分かりました。面積が大きいことの原因を特定するために、標準溶液を調査しました。まず、移管元ラボと移管先ラボの間の記録のレビューに基づいて、SOP に記載されている通り、標準溶液の濃度が 0.2% 未満であることが確認されました。明確な根本原因が分からないため、ラボで分析を繰り返し、標準溶液とサンプル溶液を再調製するように求められました。2 回目の分析の標準溶液では、ラボ 6-2 についての図 9 から分かるように、ピーク面積が他のラボと同等でした。さらに、再調製した標準溶液とサンプル溶液のデータを使用すると、不純物の割合の計算値は、他のラボから得られた値と同等でした(表 4)。したがって、不純物の割合の計算値はすべてのシステムにわたって同等になりました。これにより、標準サンプルの調製が、考えられる原因として特定されました。
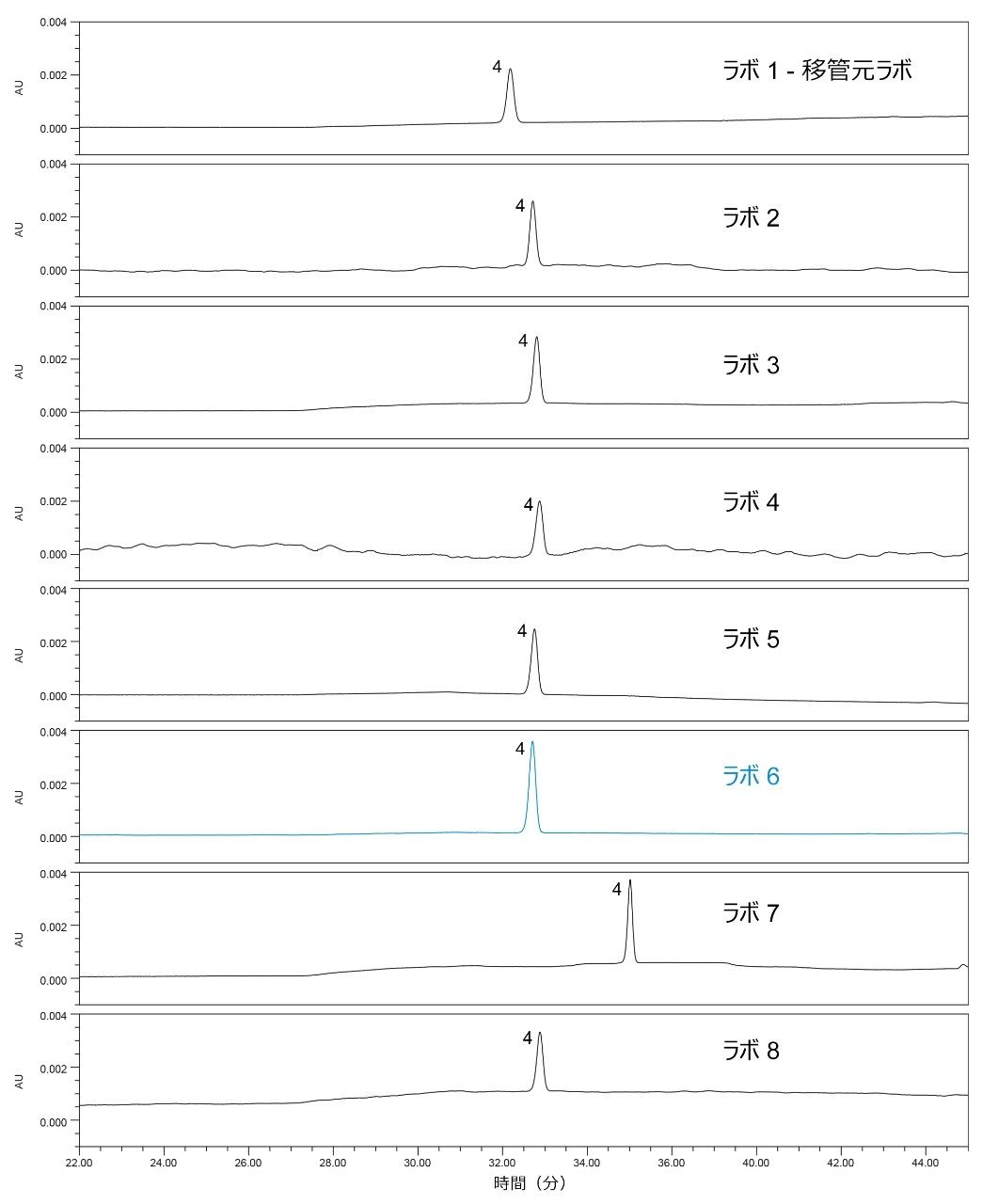 図 8. この試験に参加した 8 つのラボで得られた標準溶液の結果。ピーク 4:クエチアピン(API)。
図 8. この試験に参加した 8 つのラボで得られた標準溶液の結果。ピーク 4:クエチアピン(API)。
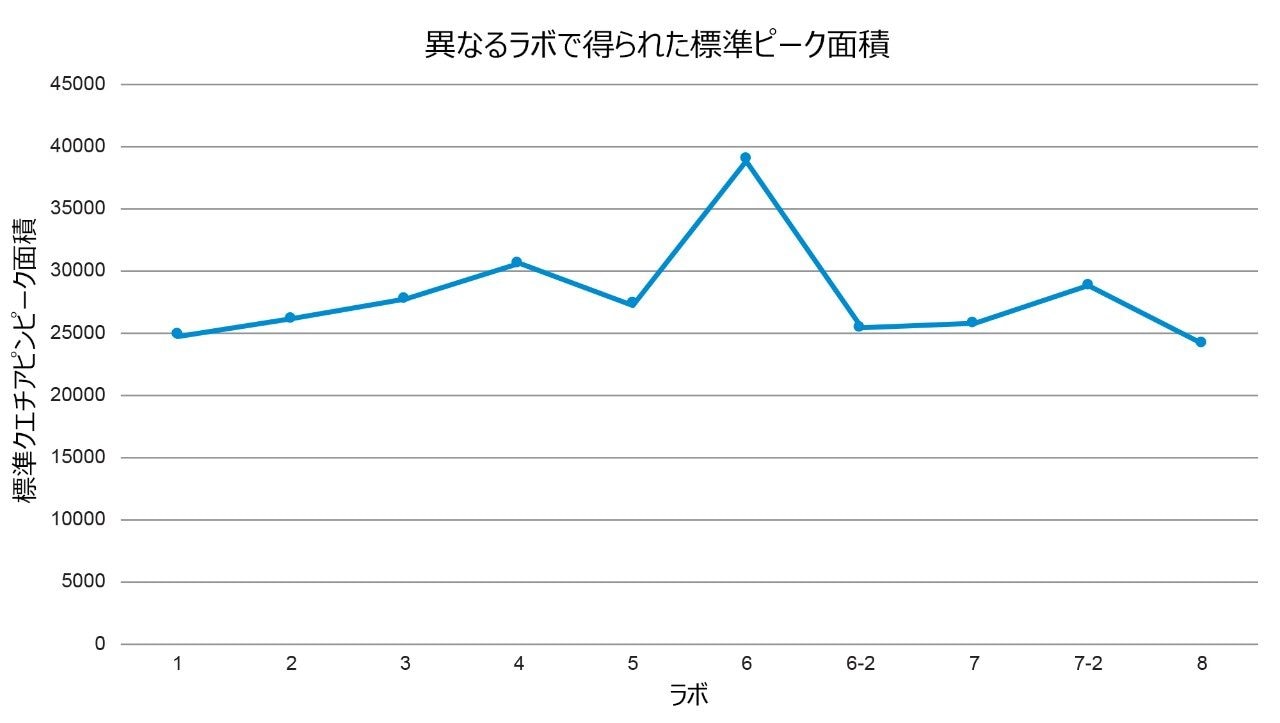 図 9. 8 つのラボすべての標準溶液のクエチアピン(API)ピークのピーク面積
図 9. 8 つのラボすべての標準溶液のクエチアピン(API)ピークのピーク面積
解決策
このエラーの解決策は、SOP 標準原液の調製法を変更して、試験で見られた変動を最小限に抑えることです。元の SOP では、単一の mg 重量が使用されていましたが、このような低重量では、サンプル調製の変動が定量結果に影響を及ぼす可能性が高まります。ストック溶液に用いる重量を増やすことにより、分析者間および実験室間の変動を減らすことができます。
結論
クエチアピンフマル酸塩の USP 不純物分析法が、Arc HPLC システムで正常にグローバルに移管でき、すべてのシステム適合性要件が満されました。結果の変動がラボ間で見られましたが、調査により、それぞれの外れ値の原因が異なることが示されました。サンプル調製に起因する変動もあれば、装置構成の違いに起因する変動もありました。一方、システム構成と分析法が結果に及ぼす影響を十分に理解することで、移管元ラボは体系的なアプローチで外れ値を調査することができました。ここに記載した試験では、以下の一般的な手順を行いました。
1. 外れ値の結果およびその潜在的原因の特定
2. 検出器やチューブ内径などのシステム構成のレビュー
3. 分析者間のサンプルおよび移動相の調製手順の違いのレビュー
このアプローチは、規格外の結果の調査にも使用できます。この試験では、分析法のリスクを理解し、管理戦略を実施することで、分析法移管を成功できることが示されました。
参考文献
- Dlugasch A, Hong P, Tran P. Successful Global Cross Lab Method Transfer of a USP Organic Impurities Method to an Arc HPLC Using a Risk-based Approach.Waters Application Note, 720007285EN, 2021.
- USP, Quetiapine Fumarate.United States Pharmacopeia and National Formulary (USP 43-NF38) 2020, (GUID-DBEED03E-7C75-4167-BD21-4E30BA2EFF2B_2_en-US), 3800.
720007376JA、2021 年 9 月