使用新型信息学工作流程进行mRNA酶解物寡核苷酸图谱分析
摘要
LC-MS和信息学技术的前沿进展为科学家设计mRNA关键属性分析的创新策略提供了支持。本应用纪要介绍了mRNA序列图谱分析从使用RNase T2酶进行样品前处理,到使用UPLC™-QTof MS进行数据采集,再到使用新型信息学软件进行数据分析的工作流程。本文详细探讨了结合使用新型RNA酶与自动化软件处理方法,对绿色荧光蛋白(GFP) mRNA进行的酶解后寡核苷酸图谱分析。结果表明,基于寡核苷酸酶解产物的唯一精确质量数,序列覆盖率得以提高。除序列确认外,我们还证明,使用新型RNase T2酶解方法对5'端加帽效率分析也有助益。
优势
- 全新信息学工作流程采用waters_connect™ MAP Sequence应用程序,简化了使用UPLC QTof MS采集数据的mRNA酶解物寡核苷酸图谱分析
- 与传统的RNase T1酶解相比,RNase T2酶(MC1和Cusativin)具有独特的切割特异性,并可生成重叠酶解产物,从而实现更高的序列覆盖率
简介
近期两种针对COVID的mRNA疫苗的研发与获批,将RNA疗法推至生物制药行业的焦点位置1~3。 由此产生的快速产品开发需求,亟需建立能够精准表征mRNA关键质量属性(CQA)(包括序列完整性与修饰完整性)的分析方法。沃特世此前已开发了两种直接评估mRNA CQA的工作流程:5'端加帽效率测定和Poly(A)尾异质性分析,这两种工作流程均使用waters_connect INTACT Mass应用程序;此外,近期还展示了一种基于MAP Sequence应用程序的新工作流程,用于100 mer单向导RNA的寡核苷酸图谱分析4,5,6。
传统的寡核苷酸测序技术(如Sanger测序和新一代测序技术(NGS))因具有成本效益和通量优势已被广泛应用于mRNA的序列分析,但其无法评估mRNA分子在合成与降解过程中产生的多种修饰。相比之下,LC-MS方法因其出色的专属性、灵敏度和定量性能正日益受到关注,能够轻松检测碱基及骨架修饰7~10。 总体而言,复杂生物制剂的表征需要采用正交技术来全面了解产品质量。
传统的LC-MS RNA酶解物寡核苷酸图谱分析工作流程繁琐耗时,涉及大量人工数据分析与整理工作。在近期发布的一篇应用纪要中,我们介绍了一种UPLC-MS和信息学工作流程,该流程通过使用一系列酶(包括RNase T1、RNase T2(RapiZyme MC1和Cusativin)以及hRNase4)分别酶解sgRNA,进行其序列的自动化图谱分析6。 本应用纪要将该工作流程(如图1所示)的应用范围扩展至采用多种RNA酶酶解的更大分子量mRNA,其中包括两种新近推出的RNase T2酶(RapiZyme MC1和RapiZyme Cusativin)。这两种酶具有独特的切割模式,可显著提高序列覆盖率6,11。 通过快速(<2分钟)自动化分配sgRNA酶解产物,并结合简化的数据处理与审查界面,该流程为sgRNA分析提供了显著优势,同时有望在更复杂的mRNA表征任务中进一步提升效率。此外,与广泛使用的RNase T1酶解方法相比,RapiZyme MC1和RapiZyme Cusativin酶解方法可对mRNA加帽效率的共测量展现出更优的结果。
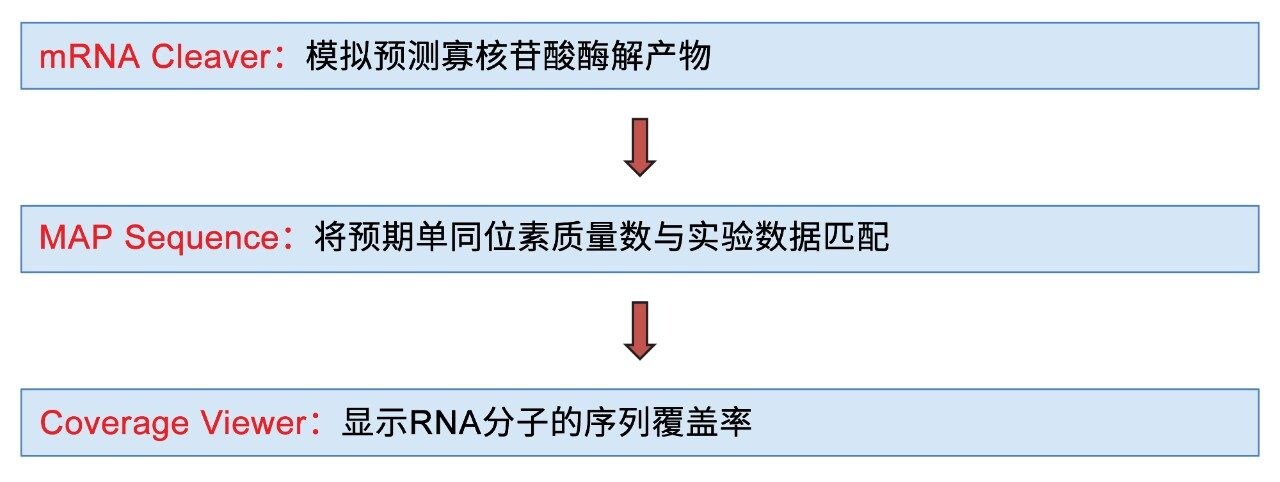 图1.使用mRNA Cleaver MicroApp、MAP Sequence应用程序和Coverage Viewer MicroApp对mRNA酶解物进行UPLC-MS处理的工作流程示意图。
图1.使用mRNA Cleaver MicroApp、MAP Sequence应用程序和Coverage Viewer MicroApp对mRNA酶解物进行UPLC-MS处理的工作流程示意图。
实验
试剂和样品前处理
二异丙基乙胺(DPA,纯度99%,产品目录号D214752–500ML)和1,1,1,3,3,3-六氟异丙醇(HFIP,纯度99%,产品目录号105228-100G)购自Millipore Sigma(美国密苏里州圣路易斯)。甲醇(LC-MS级,产品目录号34966–1L)购自Honeywell(美国卡罗来纳州夏洛特)。HPLC级I级去离子(DI)水使用Milli-Q系统(Millipore,美国马萨诸塞州贝德福德)净化。流动相每日现配现用。用于mRNA酶解的无核酸酶超纯水(产品目录号J71786.AE)购自Thermo Fisher Scientific(美国马萨诸塞州沃尔瑟姆)。
基于水母绿色荧光蛋白(GFP)序列的mRNA结构由Biosynthesis(美国德克萨斯州路易斯维尔)通过IVT(体外转录)合成方法定制而成。合成的mRNA分子具有Cap1结构(序列为:7MeGpppA(2'-OMe),元素组成:C32H42N15O26P5),其后含有1019个核苷酸,无Poly(A)尾序列。
经色谱纯化、不含动物源组分的核糖核酸酶T1(产品目录号LS01490,500 kU),从米曲霉中分离获得,购自Worthington Biochemical Corporation(美国新泽西州莱克伍德)。将冻干酶溶于5 mL 100 mM碳酸氢铵(产品目录号5.33005-50G,Millipore Sigma)中,制备100单位/μL的溶液。使用RNase T1酶解mRNA时,将5 μL 5 µM GFP mRNA与25 μL无核酸酶的水和10 μL RNase T1酶(1000单位)混合,在37 oC下酶解15分钟。在QuanRecovery™ MaxPeak™ 300 µL样品瓶中制备酶解混合物,随后立即以5 μL进样体积对酶解物进行LC-MS分析。
RapiZyme MC1(P/N:186011190,10000单位/管)和RapiZyme Cusativin(P/N:186011192,10000单位/管)是沃特世公司最近推出的两款新型RNA酶6,7。 RapiZyme MC1和Cusativin的GFP mRNA酶解方案非常相似。使用RapiZyme MC1时,将GFP mRNA(10 μL,5 µM溶液)置于含有200 mM乙酸铵的缓冲液(pH 8.0)中,在90 oC下处理2分钟使其变性。使用RapiZyme Cusativin时,将mRNA(10 μL,5 µM溶液)置于含有200 mM乙酸铵的缓冲液(pH 9.0)中,在90 oC下处理2分钟使其变性。两种样品均置于冰上冷却,然后用微量离心机处理以收集样品液滴。加入50单位的酶(RapiZyme MC1或RapiZyme Cusativin各为1 μL)和8 μL无核酸酶的水,使最终体积约20 μL,然后在Eppendorf恒温混合器中在37 oC下将mRNA酶解60分钟。将溶液加热至70 ºC并保持15分钟,使酶失活,从而终止酶解。随后立即以5 μL进样体积对酶解物进行LC-MS分析。
所有数据集均使用waters_connect UNIFI™应用程序3.6.0.21版采集,随后使用MAP Sequence应用程序在mRNA Cleaver和Coverage Viewer MicroApps的协助下进行处理。
液相色谱条件
|
LC-MS系统: |
Xevo™ G3 QTof与ACQUITY™ Premier UPLC(二元)系统联用的LC-MS系统 |
|
色谱柱: |
ACQUITY Premier BEH™ C18寡核苷酸分析专用柱130 Å, 1.7 µm, 2.1 x 150 mm(P/N:186009486) |
|
柱温: |
60 oC |
|
流速: |
300 µL/min |
|
流动相: |
溶剂A:10 mM DPA(二异丙基乙胺),40 mM HFIP(1,1,1,3,3,3-六氟异丙醇),溶于去离子水中,pH 8.5 溶剂B:10 mM DPA,40 mM HFIP,溶于50%乙醇中 |
|
样品温度: |
8 oC |
|
样品瓶: |
QuanRecovery MaxPeak HPS样品瓶(P/N:186009186) |
|
进样体积: |
5 µL |
|
清洗溶剂: |
灌注溶剂:50%甲醇 样品管理器清洗溶剂:50%甲醇 密封清洗液:含20%乙腈的去离子水溶液 |
梯度表

质谱条件
|
质谱系统: |
Xevo™ G3 QTof质谱仪 |
|
电离模式: |
ESI(-) |
|
采集模式: |
MSE |
|
采集速率: |
1 Hz |
|
毛细管电压: |
2.5 kV |
|
锥孔电压: |
40 V |
|
离子源补偿: |
60 V |
|
离子源温度: |
120 oC |
|
脱溶剂气温度: |
550 oC |
|
锥孔气流速: |
50 L/h |
|
脱溶剂气流速: |
600 L/h |
|
TOF质量范围: |
340~4000(MSE采集) |
|
低能量CE: |
6 V |
|
高能量CE梯度: |
25~50 V |
|
实时校正标准液: |
50 pg/μL亮氨酸脑啡肽 |
|
数据采集: |
waters_connect 3.6.0.21 |
|
数据处理: |
waters_connect 3.6.0.21 |
|
数据处理: |
mRNA Cleaver MicroApp 1.1.0版 |
结果与讨论
随着生物治疗机构不断改进产品开发流程,LC-MS方法在RNA序列分析中的应用愈加广泛,这项技术有助于实现更全面的初始表征和持续可靠的序列确认。色谱重现性和分离度的提升、高分辨率MS的易用性增强,以及MS系统灵敏度和准确度的改进,使得序列确认和碱基修饰检测更加可靠。最重要的是,新型寡核苷酸信息学工具使RNA图谱数据分析这一艰巨任务变得更快捷、更简单。
虽然LC-MS用于生物制药蛋白的酶解肽图分析已十分常规,但将类似方法应用于RNA仍更具挑战性。与由20种不同氨基酸组成的蛋白质不同,mRNA仅含有四种构成单元(A、C、G、T),这使得生成具有唯一质量数可对mRNA进行图谱分析的寡核苷酸酶解产物更加困难。高频率切割酶(如具有G-切割特异性的RNase T1)会产生大量同分异构体甚至完全相同的mRNA酶解产物,导致序列分配模糊。而低频率酶切位点的酶能够产生更长的酶解产物,尤其是在生成部分漏切寡核苷酸的情况下。本研究中采用的新型RNase 2型酶(RapiZyme MC1和RapiZyme Cusativin)更有可能产生具有唯一质量数的可分配酶解产物。
本研究设计了一套信息学工作流程(图1),采用waters_connect MAP Sequence应用程序进行数据处理和序列分配,有利于对酶解和UPLC-MS数据采集后采集的LC-MS数据集进行快速、自动化的数据处理6。 第一步,使用mRNA Cleaver MicroApp生成指定mRNA分子的预测模拟酶解寡核苷酸产物。第二步,waters_connect MAP Sequence应用程序处理UPLC-MS数据,并将预测的酶解寡核苷酸中性单同位素质量数与实验MS1数据匹配。最后一步,使用Coverage Viewer MicroApp汇总并查看mRNA酶解物的序列覆盖率。
由于该工作流程利用RNA酶解产物的精确质量数测量来进行寡核苷酸图谱分析,因此通过酶解生成明确、独特的产物,以减少对MS2碎片离子数据用户干预的依赖,对解决分配不明确的问题至关重要。RapiZyme MC1和RapiZyme Cusativin酶具有独特的切割特异性,还可以通过调节酶量和酶解时间刻意产生漏切,生成更长的独特酶解产物。本研究使用非数据依赖型MSE采集确认寡核苷酸酶解产物的分配结果。未来版本的应用程序中将纳入高能量MSE碎片离子信息,特别用于解决一些残留的模糊分配问题。
mRNA寡核苷酸图谱分析
本研究使用三种不同的核糖核酸酶(RNase T1、RapiZyme MC1和RapiZyme Cusativin)酶解GFP mRNA样品,采集了三个独立的LC-MSE寡核苷酸图谱数据集。使用MAP Sequence应用程序处理这三个UPLC-MSE数据集,参数如图2所示,并将结果导出至Coverage Viewer MicroApp以快速查看序列覆盖率。图3A、4A和5A分别展示了RNase T1、RapiZyme MC1和RapiZyme Cusativin的TIC色谱图。据了解,与RNase T1相比,MC1和Cusativin会产生明显更多的漏切,这一酶解特征清楚地反映在图4A和5A所示的复杂色谱图中6~9。
序列图谱分析结果的比较(图3B、4B和5B)表明,RNase T1的模糊分配酶解产物数量更高,因此其独特覆盖率显著较低(约60%,图3B),而MC1(约97%,图4B)和Cusativin(约85%,图5B)的覆盖率较高。导致这一结果的直接原因是,RNase T1的切割特异性更广(在每个G残基后切割),而MC1和Cusativin的切割特异性依赖于高度特异的二核苷酸基序6~9。 MC1在尿苷残基的5'端切割,有三个主要酶切位点(A_U / C_U / U_U)和两个次要酶切位点(C_A / C_G)。Cusativin在胞苷残基的3'-端切割,有四个主要酶切位点(C_A / C_G / C_U / U_A)和三个次要酶切位点(A_U / G_U / U_U)。RNase T1会在其所有酶解产物的3'端添加一个线性磷酸基团,而MC1和Cusativin产生的酶解产物主要以3'环状磷酸形式存在。
与RNase T1相比,MC1和Cusativin产生受控漏切的能力也更高,因此在mRNA Cleaver预测工具中,这两种酶允许最多4个漏切,而RNase T1只允许2个漏切。由于漏切数量更多,MC1和Cusativin产生的酶解产物相比RNase T1更长。这些更长的寡核苷酸产物更可能具有唯一质量数,从而降低出现模糊分配的可能性。得益于这些酶解特征,MC1和Cusativin的图谱分析结果表现出更高的覆盖率。
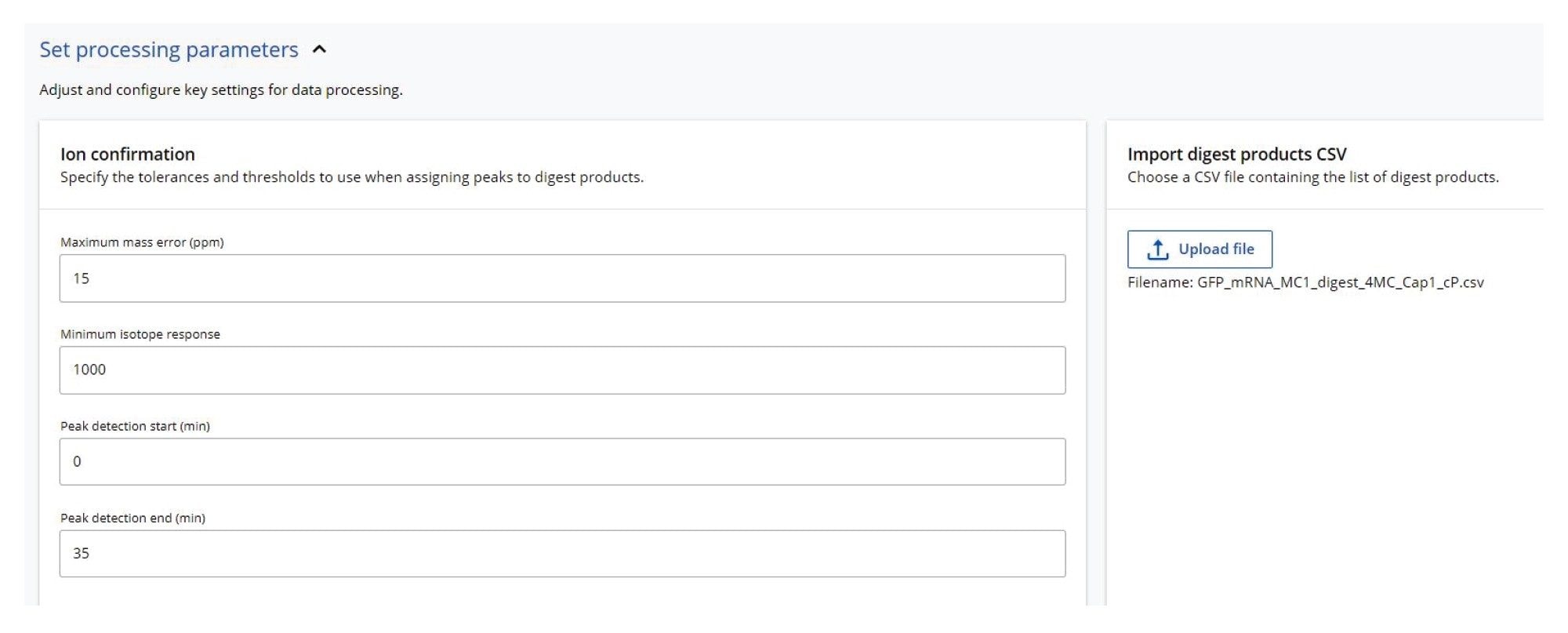 图2.MAP Sequence处理参数。此应用程序使用mRNA Cleaver模拟预测的酶解产物(参见右侧的csv文件)与酶解样品的UPLC-MS数据集匹配。
图2.MAP Sequence处理参数。此应用程序使用mRNA Cleaver模拟预测的酶解产物(参见右侧的csv文件)与酶解样品的UPLC-MS数据集匹配。
 图3. (A) RNase T1酶解GFP mRNA的TIC色谱图;(B) Coverage Viewer MicroApp的结果,汇总并显示了RNase T1酶解分析后分配的寡核苷酸独特序列覆盖率。
图3. (A) RNase T1酶解GFP mRNA的TIC色谱图;(B) Coverage Viewer MicroApp的结果,汇总并显示了RNase T1酶解分析后分配的寡核苷酸独特序列覆盖率。
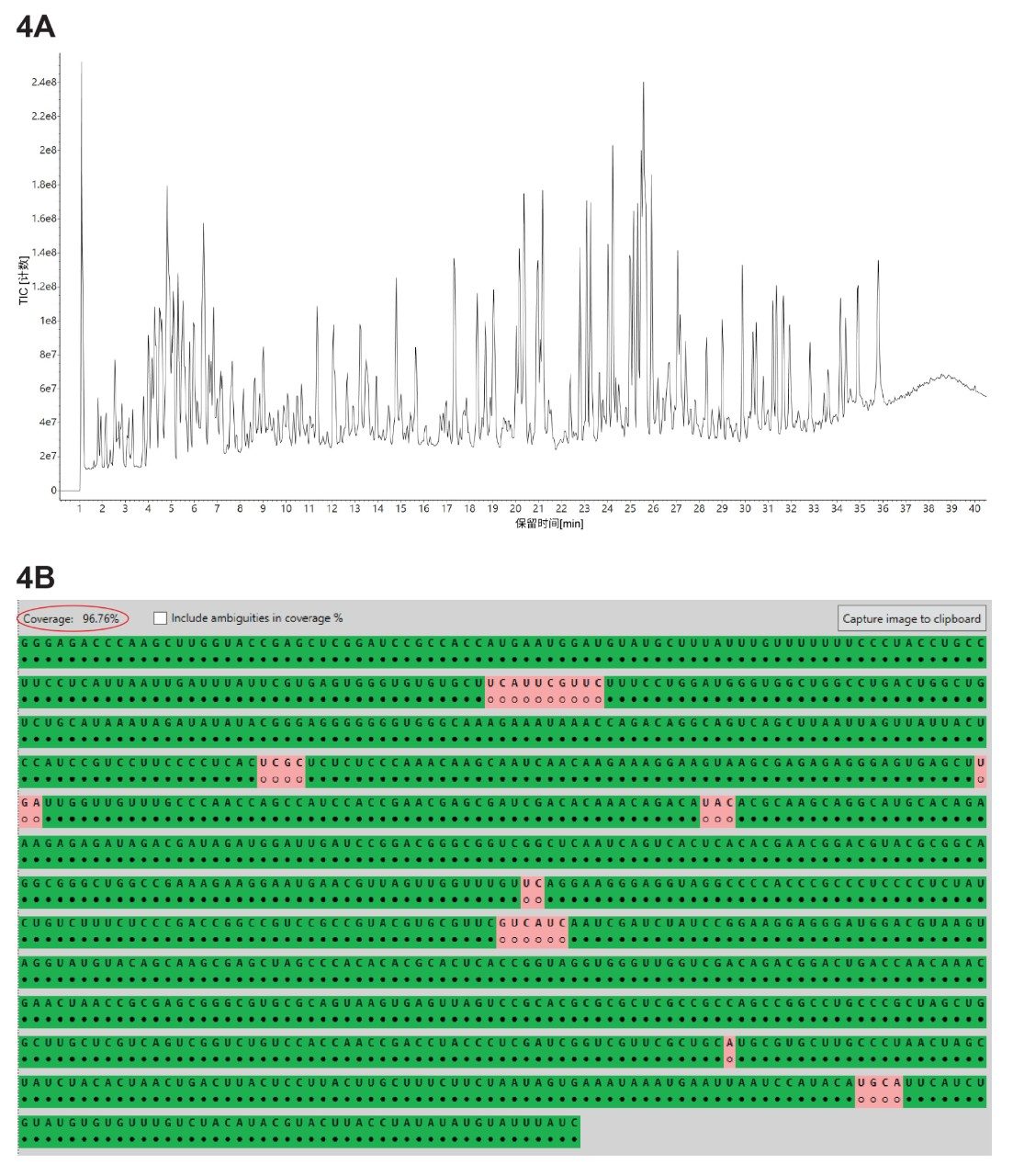 图4. (A) RapiZyme MC1酶解GFP mRNA的TIC色谱图;(B) Coverage Viewer MicroApp的结果,汇总并显示了RapiZyme MC1酶解分析后分配的寡核苷酸独特序列覆盖率。
图4. (A) RapiZyme MC1酶解GFP mRNA的TIC色谱图;(B) Coverage Viewer MicroApp的结果,汇总并显示了RapiZyme MC1酶解分析后分配的寡核苷酸独特序列覆盖率。
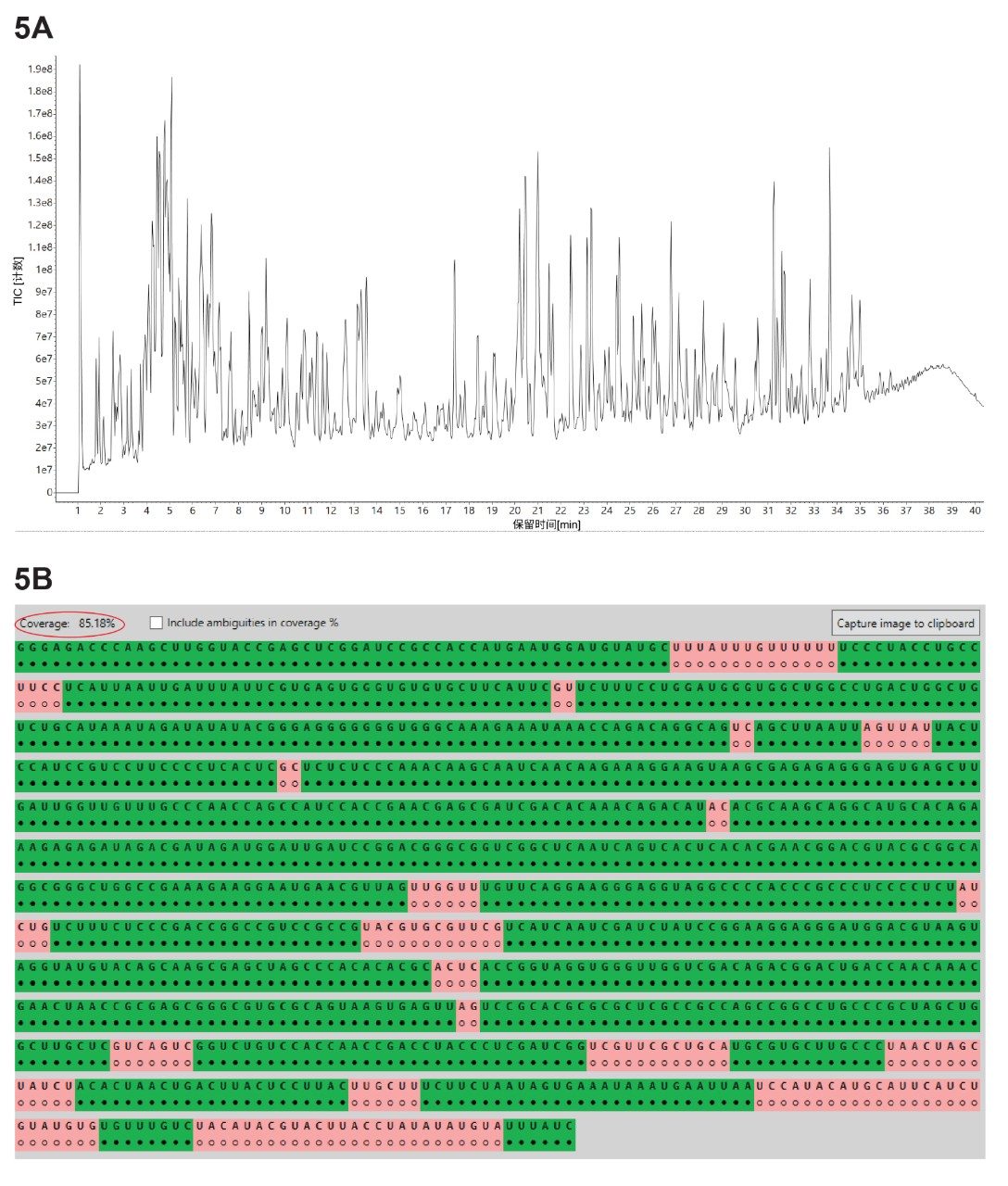 图5.(A) RapiZyme Cusativin酶解GFP mRNA的TIC色谱图;(B) Coverage Viewer MicroApp的结果,汇总了RapiZyme Cusativin酶解分析后分配的寡核苷酸独特序列覆盖率。
图5.(A) RapiZyme Cusativin酶解GFP mRNA的TIC色谱图;(B) Coverage Viewer MicroApp的结果,汇总了RapiZyme Cusativin酶解分析后分配的寡核苷酸独特序列覆盖率。
如果序列分配模糊,可以使用waters_connect中的CONFIRM Sequence应用程序进一步研究高能量MSE数据,将碎片离子分配给可能性最高的寡核苷酸序列12。为阐明此方法,我们选择了一对由RNase T1酶解GFP mRNA产生的16 mer酶解产物。这对结构异构体为U251:G266(序列UCC UUC CCC UCA CUC G)和C555:G570 (CCC UCC CCU CUA UCU G),二者的核苷酸组成相同。如插图(图6顶部)所示,MAP Sequence自动基于多个母离子(电荷态2~9)找到这两个序列,并将它们标记为“替代产物分配”(alternative product assignments)。图6还显示了相应三电荷母离子(m/z=1654.86)的提取质量色谱图,指示存在一个高丰度色谱峰,该峰可能属于一种或两种寡核苷酸产物。使用CONFIRM Sequence自动分配在该寡核苷酸酶解产物的高能量ESI-MSE谱图中(图7)检测到的碎片离子,可靠地揭示C555:G570序列的匹配,序列覆盖率达到100%。
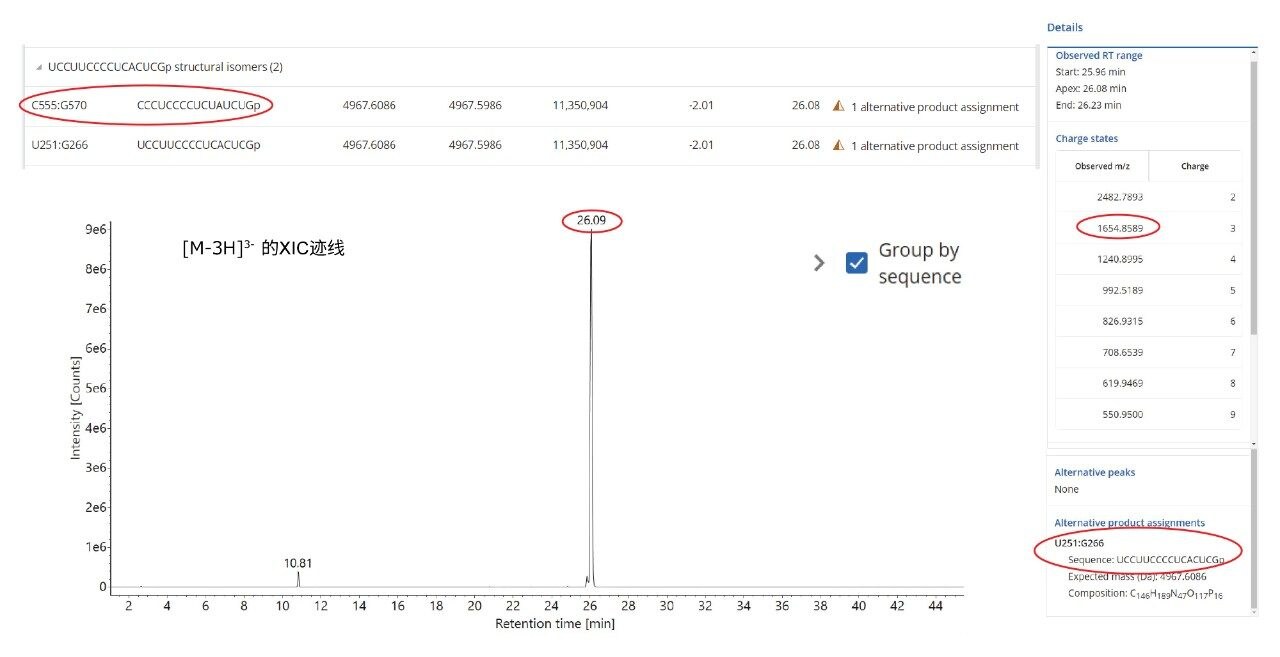 图6.三电荷母离子(m/z 1654.86)的提取质量色谱图,分配模糊,可能会被分配给RNase T1切割GFP mRNA产生的同分异构酶解产物U251:G266或C555:G570。顶部插图显示了MAP Sequence对这两种结构异构体的分配结果,右侧插图显示了MAP Sequence在该寡核苷酸产物的ESI-MS谱图中检测到的电荷态(2–9)。
图6.三电荷母离子(m/z 1654.86)的提取质量色谱图,分配模糊,可能会被分配给RNase T1切割GFP mRNA产生的同分异构酶解产物U251:G266或C555:G570。顶部插图显示了MAP Sequence对这两种结构异构体的分配结果,右侧插图显示了MAP Sequence在该寡核苷酸产物的ESI-MS谱图中检测到的电荷态(2–9)。
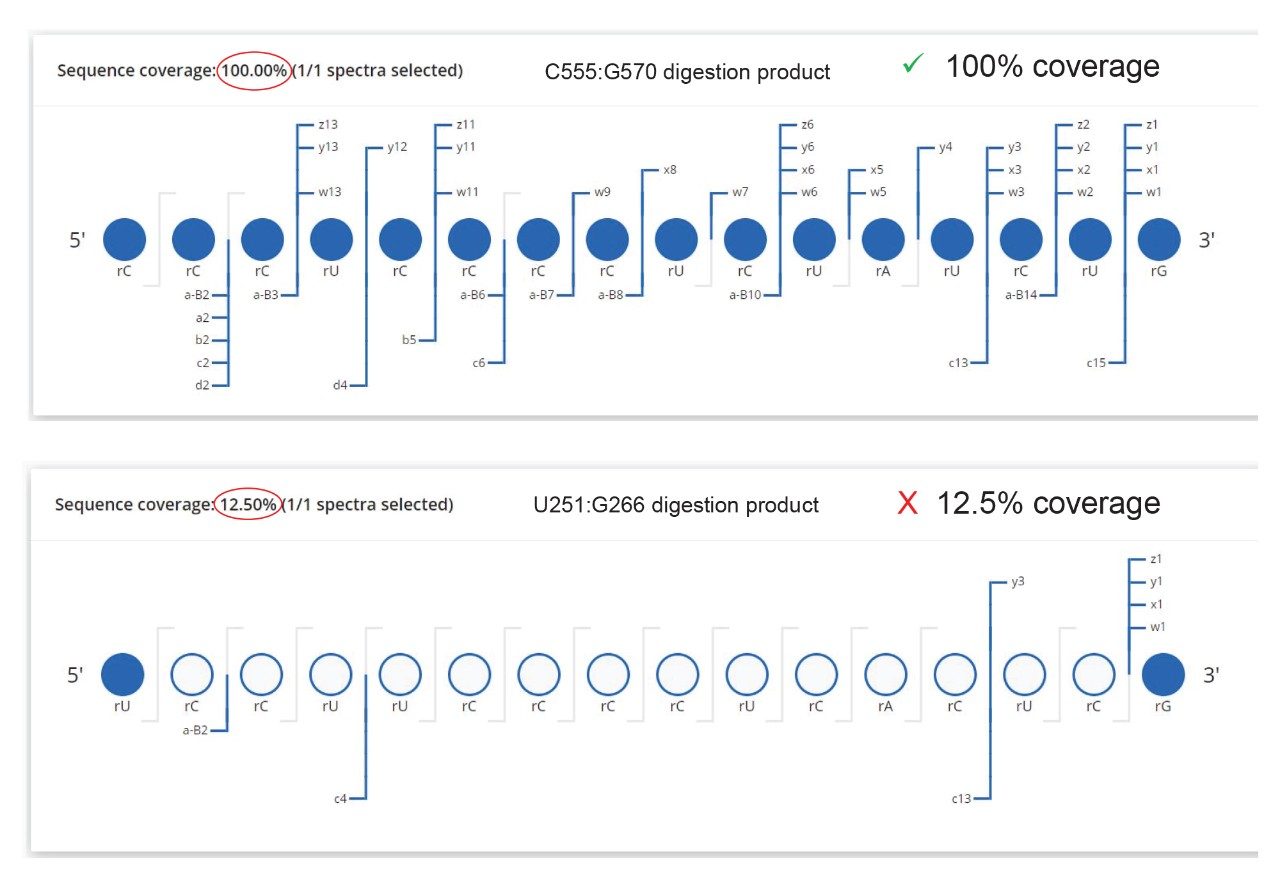 图7.CONFIRM Sequence屏幕截图,显示了RNase T1酶解GFP mRNA产生的两种可能的16 mer酶解产物的序列覆盖率点图。图6中26.09 min处检测到的色谱峰为C555:G270 16 mer寡核苷酸(CCC UCC CCU CUA UCU G),序列覆盖率达到100%。只有少数末端碎片离子可以分配给另一个同分异构序列(UCC UUC CCC UCA CUC G)。
图7.CONFIRM Sequence屏幕截图,显示了RNase T1酶解GFP mRNA产生的两种可能的16 mer酶解产物的序列覆盖率点图。图6中26.09 min处检测到的色谱峰为C555:G270 16 mer寡核苷酸(CCC UCC CCU CUA UCU G),序列覆盖率达到100%。只有少数末端碎片离子可以分配给另一个同分异构序列(UCC UUC CCC UCA CUC G)。
沃特世之前发布的应用纪要中介绍了几个mRNA加帽分析示例5,13。mRNA帽状结构位于mRNA的5'端,通常由一个完全成熟的7-甲基鸟苷通过三磷酸酯键以5'-5'连接方式与mRNA结合(m7Gppp)。对于GFP mRNA,我们使用了一种Cap1结构变体,即腺苷残基经2'-O-甲基化修饰后再连接到mRNA上。因此,GFP mRNA的Cap1结构用m7GpppAm表示。
未加帽和加帽的GFP 5' mRNA酶解产物均在MC1 mRNA酶解物中检出,如图8的提取质量色谱图所示。经MC1酶解后,未加帽版本(m/z=1419.5302,-3)在22.0 min处洗脱(上图,图8A),Cap1修饰版本(m/z=1700.8962,-3)在24.7 min处洗脱(下图,图8B)。使用这些三电荷态XIC峰的积分峰面积,计算出加帽效率为79.8%。
根据检测到的最高丰度电荷态([M-3H]3-)的同位素分布(图9),进一步鉴定这些寡核苷酸物质。图8A突出显示了序列为GGA AGA CCC AAG C的游离(未加帽)13 mer MC1酶解寡核苷酸的单同位素峰,下图(图8B)显示了Cap1修饰的13 mer寡核苷酸的单同位素峰。在本例中,MC1和Cusativin在切割GFP mRNA的5'端后产生了相同的酶解产物(序列GGA AGA CCC AAG C),从而能够测量mRNA加帽效率。但在RNase T1酶解案例中,由于5'端的第一个mRNA残基是鸟苷,导致未加帽的酶解产物过短(仅一个核苷酸),在所用的IP-RP条件下无法保留。因此,只能检测到Cap1酶解产物(序列m7GpppAmGp,最高丰度单同位素离子:m/z=602.5482,-2,数据未显示),并且无法测量mRNA加帽效率。因此,对于GFP mRNA,只有MC1和Cusativin可以成功用于计算加帽效率。
这些结果突显了质谱技术目前在大分子mRNA序列确认中的实用性,但工作流程效率和自动化仍有提升空间,且仍是活跃的研究方向。未来需持续开发处理此类复杂数据集的自动化软件,并扩展酶的工具库,以进一步提升这一新兴方法的实用性。
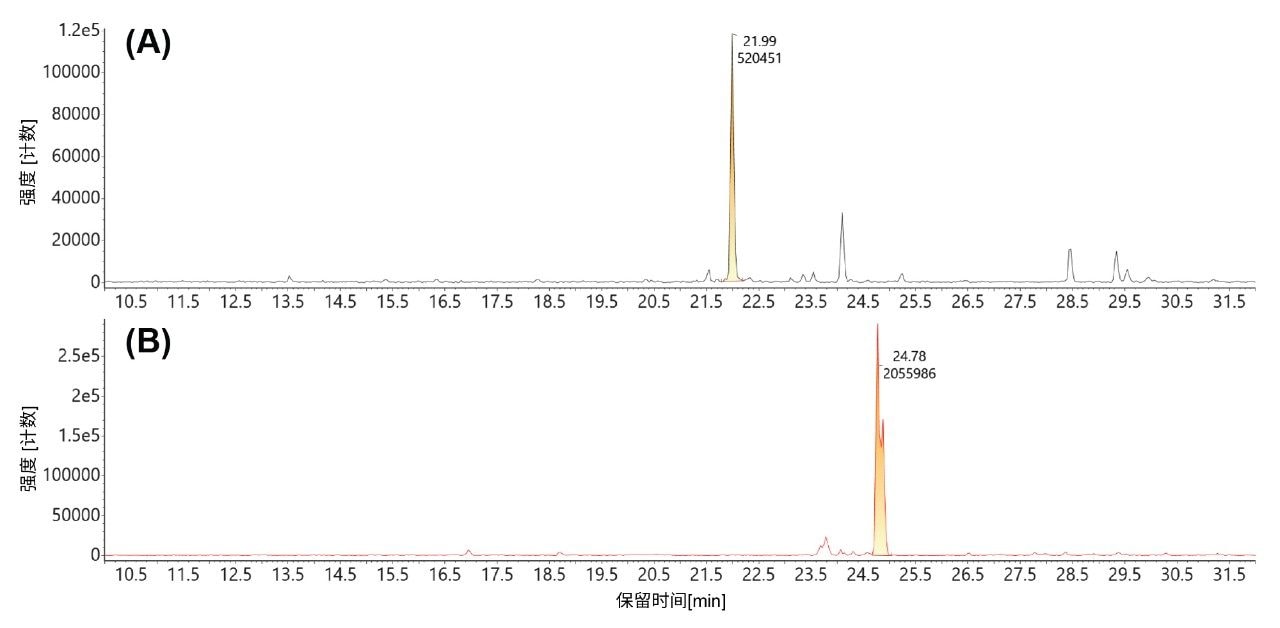 图8.MC1酶解产生的同一种13 mer酶解产物(GGA AGA CCC AAG C)未加帽(A)和加帽(B)版三电荷母离子的提取质量色谱图:(A) m/z=1419.51 (-3)未加帽的GGA AGA CCC AAG C寡核苷酸;(B) m/z =1700.87 (-3) Cap1修饰的GGA AGA CCC AAG C寡核苷酸。
图8.MC1酶解产生的同一种13 mer酶解产物(GGA AGA CCC AAG C)未加帽(A)和加帽(B)版三电荷母离子的提取质量色谱图:(A) m/z=1419.51 (-3)未加帽的GGA AGA CCC AAG C寡核苷酸;(B) m/z =1700.87 (-3) Cap1修饰的GGA AGA CCC AAG C寡核苷酸。
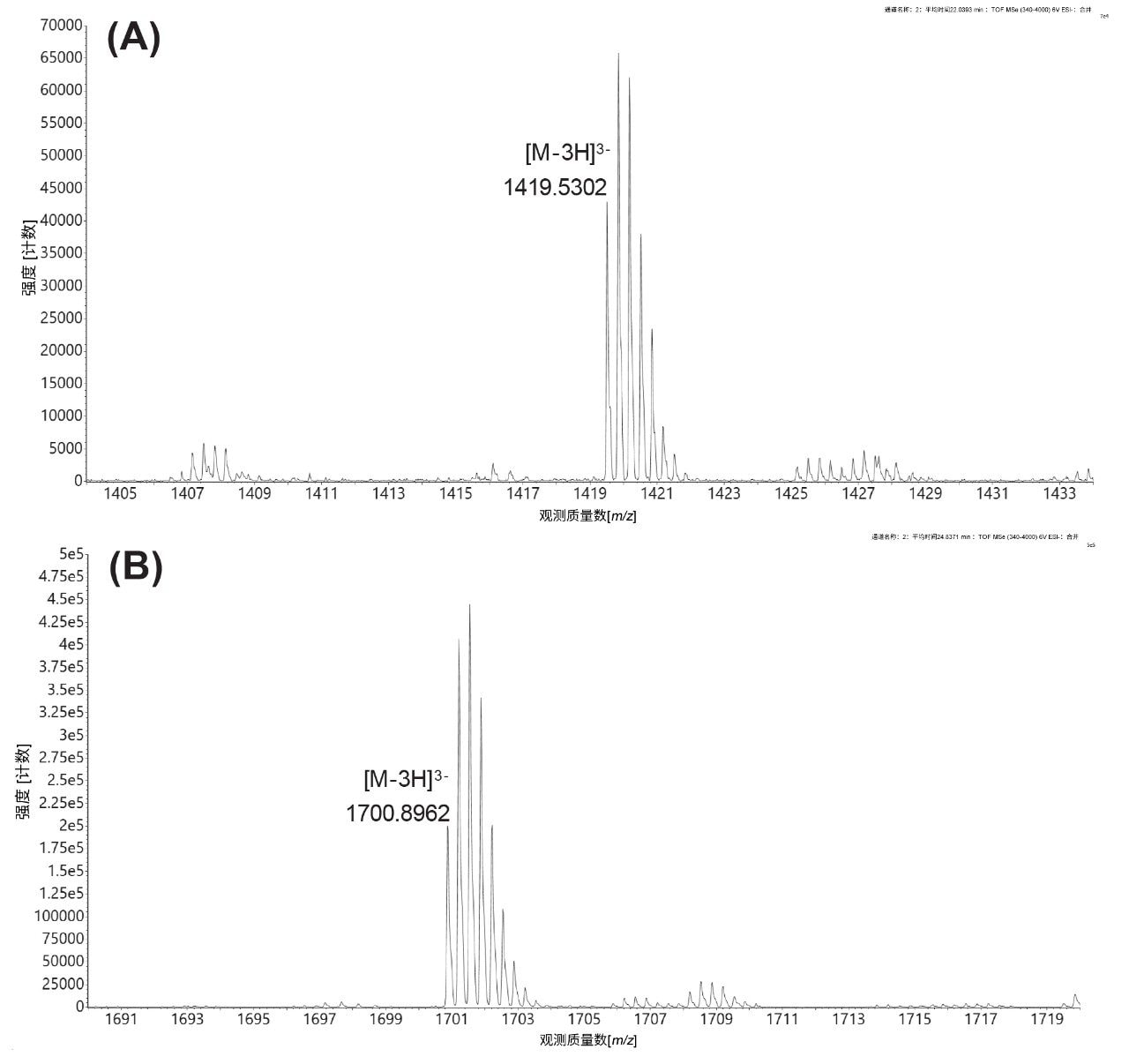 图9.MC1酶解GFP mRNA后产生的未加帽(A)和Cap1修饰(B)版13 mer寡核苷酸(序列为GGA AGA CCC AAG C)的三电荷离子的同位素分布。在相应的ESI-MS谱图中标记了未加帽和加帽寡核苷酸的单同位素峰。
图9.MC1酶解GFP mRNA后产生的未加帽(A)和Cap1修饰(B)版13 mer寡核苷酸(序列为GGA AGA CCC AAG C)的三电荷离子的同位素分布。在相应的ESI-MS谱图中标记了未加帽和加帽寡核苷酸的单同位素峰。
结论
- 本研究展示了一套全新的信息学工作流程,该流程采用沃特世的waters_connect MAP Sequence应用程序,可利用UPLC-MS采集的数据对mRNA酶解物进行MS1寡核苷酸图谱分析。
- 与RNase T1相比,两种新型的RNase T2酶(RapiZyme MC1和RapiZyme Cusativin)具有独特的酶解特异性,能够生成更多的完全酶解寡核苷酸和漏切寡核苷酸,从而实现更高、更可靠的重叠序列覆盖率。
- 使用waters_connect CONFIRM Sequence应用程序将高能量碎片离子与同分异构寡核苷酸序列匹配,从而区分寡核苷酸酶解产物的同分异构体
参考资料
- Xu S, Yang K, Li R, Zhang L. mRNA Vaccine Era-Mechanisms, Drug Platform and Clinical Prospection, Intl J Mol Sci Chem, 2020, 21 (18), 6582, doi: 10.3390/ijms21186582.
- Verbeke R, Lentacker I, De Smedt SC, Dewitte H. The dawn of mRNA vaccines: The COVID-19 case.J Controlled Release, 2021, 333, 511–520, doi: 10.1016/ j.jconrel.2021.03.043.
- Jackson NA, Kester KE, Casimiro D, Gurunathan S, DeRosa F. The Promise of mRNA Vaccines: A Biotech and Industrial Perspective.npj Vaccines, 2020, 5, https://doi.org/10.1038/s41541-020-0159-8.
- 使用离子对液相色谱和质谱法进行合成mRNA寡核苷酸图谱分析, 2022, 沃特世应用纪要.2022年6月. 720007669ZH.
- 使用BioAccord LC-MS系统和INTACT Mass waters_connect应用程序进行RNA CQA分析, 2023, 沃特世应用纪要.2023年11月. 720008130ZH.
- RNA Digestion Product Mapping Using an Integrated UPLC-MS and Informatics Workflow, 2024, Waters application note. September, 2024. 720008553.
- Jiang T, Yu N, Kim J, Murgo JR, Kissai M, Ravichandran K, Miracco E, Presnyak V, Hua S. Oligonucleotide Sequence Mapping of Large Therapeutic mRNAs via Parallel Ribonuclease Digestions and LC-MS/MS, Anal Chem, 2019, 91, 8500–8506, doi:10.1021/acs.analchem.9b01664.
- Vanhinsbergh CJ, Criscuolo A, Sutton JN, Keely M, Williamson AJK, Cook K, Dickman M. Characterization and Sequence Mapping of Large RNA and mRNA Therapeutics using Mass Spectrometry, Anal Chem, 2022, 94, 7339–7349, doi:10.1021/acs.analchem.2c00765.
- Gau B, Dawdy AW, Wang HL, Bare B, Castaneda CH, Friese OV, Thompson MS, Lerch TF, Cirelli DJ, Rouse JC.Oligonucleotide Mapping via Mass Spectrometry to Enable Comprehensive Primary Structure Characterization of an mRNA Vaccine Against SARS CoV-2, Sci Rep, 2023, 13, No 9038, doi:10.1038/s41598-023-36193-2.
- Tang S, Liu GY, Yan Y, Wang S, Li N. Development of a Flow Through-Based Limited Digestion Approach for High-Throughput and High-Sequence Coverage Mapping of Therapeutic mRNAs, Anal Chem, 2024, 96, 16944–17003, doi:10.1021/acs.analchem.4c04384.
- Tunable Digestion of RNA Using RapiZyme RNases to Confirm Sequence and Map Modifications, 2024, Waters application note. September, 2024. 720008539.
- CONFIRM Sequence:一款用于合成寡核苷酸及其杂质测序的waters_connect应用程序, 2022, 沃特世应用纪要.2022年7月. 720007677ZH.
- 使用离子对反相液相色谱法和BioAccord LC-MS系统快速分析合成的mRNA帽状结构, 2021, 沃特世应用纪要.2021年8月. 720007329ZH.
720008677ZH,2025年1月