穀物食品に含まれる 50 種のマイコトキシンおよびトロパンアルカロイドのためのマルチトキシン UPLC-MS/MS 分析法の開発
要約
このアプリケーションノートでは、穀物ベースの食品に含まれる 50 種の規制対象の新規マイコトキシン、アトロピン、スコポラミンについてのマルチトキシン UPLC-MS/MS 分析法の性能を説明します。高感度タンデム四重極質量分析計 Xevo TQ-XS を ACQUITY UPLC システムと組み合わせて使用し、非常に低い検出限界および定量限界を達成しました。単純な「希釈して注入」プロトコルを使用して、クリーンアップや内部標準を使用せずに、穀粉混合物を抽出しました。溶媒標準およびスパイクした抽出物を使用して検量線をプロットしました(マトリックスマッチドキャリブレーション)。検出限界および定量限界が、規制限度への準拠の確認および他の毒素レベルの調査に適していることが示されました。分析法の定量限界(m-LOQ)が最も低いのはアフラトキシンでした(0.1 µg/kg)。キャリブレーション範囲は許容範囲内であり、分析種のほとんどについて 3 桁の範囲がカバーされていました。また、すべての化合物についてマトリックス効果を計算したところ、有意であることが分かり、マトリックスマッチドキャリブレーションが必要であることがわかりました。この分析法は、SANTE ガイドラインで設定されているマイコトキシンの基準を満たしていました。 データを、定量ソフトウェアのための新しい waters_connect にインポートし、MS Quan アプリで解析することで、データ解析およびレビューの効率が改善しました。
アプリケーションのメリット
- 単一の LC-MS/MS 分析法による 50 種以上のマイコトキシンおよび植物毒素の同時測定
- 高感度 Xevo TQ-XS を使用することで、記録上最低の検出限界を達成
- 卓越した感度および優れた直線性と再現性が得られ、規制要件に適合
- 定量ソフトウェアのための新しい waters_connect を用いる MS Quan アプリケーションにより、データ解析および結果のレビューにかかる時間が短縮
はじめに
マイコトキシンは様々なカビが産生する天然に存在する二次代謝産物です。これらの化合物は、ヒトおよび動物に対して毒性があります。通常、マイコトキシンは菌類によって産生され、その親構造は多くの場合、菌自体によって修飾され、構造的に関連した化合物が多数放出されます。感染が成立すると、これらの物質は、真菌類の宿主植物によって更に修飾されます。生きた植物により、毒物の化学構造が変化し、いわゆるマスクドマイコトキシンが産生されることがあります。これらのマスクドマイコトキシンは植物に対する毒性が低いため、その生成は、作物の主要な解毒戦略です。食品・飼料の安全性を確保するため、多くの国で作物中のマイコトキシンの規制限界が設けられています。このような規制が適用されるのは、いくつかの既知のマイコトキシン(アフラトキシン(B1、B2、G1、G2、M1)、フモニシン(B1、B2、B3)、オクラトキシン A、デオキシニバレノール、ゼアラレノン、HT-2 トキシン、T-2 トキシン、トロパンアルカロイド、麦角アルカロイドなど)に限定されています1。
現在進行中の試験により、気候変動に適応させる植物育種の取組みと同様に、新しく発見されたマイコトキシン代謝物についての情報が絶え間なく得られています。法制化の準備の一環として、必要になる場合に備えて、リスク評価試験が現在実施されています。これらの化合物はしばしば「新規マイコトキシン」と呼ばれます2。 「マスクドマイコトキシン」という用語は元々、結合型マイコトキシン代謝物を指していましたが、このグループは現在ではより正確に「修飾マイコトキシン」と呼ばれ、親マイコトキシンの代謝物が含まれます3。 新規マイコトキシンおよび修飾マイコトキシンのいずれについても、その発生のエビデンスが急速に増加しており、穀物およびその他の食品/飼料中に高頻度、場合によっては高濃度で存在する可能性が実証されています4。
新規マイコトキシンや修飾マイコトキシンの一部や植物毒素は既に規制されており、将来のある段階で更なる措置が取られる可能性があります。過去 10 年間で、これらの 3 種の毒素を検査するニーズが大幅に増加し、その一部は食品/飼料業界が適正評価のために行いますが、発生に関するデータを求める声に応えるものとしても行われています。そのため、分析の適用範囲を広げ、これらの化合物を含める必要があります。この文書では、高感度 Xevo TQ-XS を使用した、穀物ベースの食品分析のためのマルチトキシン定量分析法とするため、以前のアプリケーションノート5 の適用範囲を拡張した場合の性能について記載します。
実験方法
サンプル抽出
以前の報告の手順に基づいてコムギ、オオムギ、コメ、トウモロコシの穀粉混合物を抽出しました5,6。 簡単に説明すると、5.0 g の均質化したサンプルを 50 mL のプラスチック遠心分離チューブに入れ、0.75% 酢酸および 0.2% ギ酸(v/v)を含む 20 mL の 80:20 MeCN:H2O で、10 分間の自動ボルテックス混合により抽出しました。>5000 g で 6 分間遠心分離した後、150 µL の上清を LC バイアルに移し、続いて 1,350 µL の水を加えて(1:10 希釈)ろ過(グラスファイバーシリンジフィルター)した後、LC-MS/MS を行いました。全体の希釈率は 40 倍です。
キャリブレーション標準試料の調製
以下の 2 シリーズのキャリブラントを調製しました。
- ストック混合液の連続希釈により溶媒ベースの標準を調製しました。この時、溶媒の組成は H2O:MeCN 95:5(v/v)に保ちました。
- ブランクサンプル抽出物によるストック混合液の連続希釈により、同じ範囲にわたるマトリックスマッチド標準を調製しました。
LC 条件
|
クロマトグラフィーシステム: |
ACQUITY UPLC I-Class PLUS(バイナリーソルベントマネージャー搭載) |
|
オートサンプラーおよびインジェクター: |
フロースルーニードルインジェクター(FTN)、ニードルサイズ 15 µL |
|
カラム: |
ACQUITY UPLC BEH C18(2.1 × 100 mm、粒子径 1.7 μm、ポアサイズ 130 Å、製品番号 186002352) |
|
水系移動相: |
0.5% 酢酸 および 0.1% ギ酸(v/v)を含む 1 mM 酢酸アンモニウム水溶液 |
|
有機移動相: |
メタノール + 0.5% 酢酸 + 0.1% ギ酸(v/v) |
|
ニードル洗浄溶媒: |
水:メタノール:アセトニトリル:イソプロパノール:アセトン、20:20:20:20:20 および 1% ギ酸(体積比) |
|
シール洗浄溶媒: |
水:アセトニトリル 80:20(v/v) |
|
カラム温度: |
40 ℃ |
|
サンプル温度: |
15 ℃ |
|
注入量: |
5 μL |
|
流速: |
0.40 mL/分 |
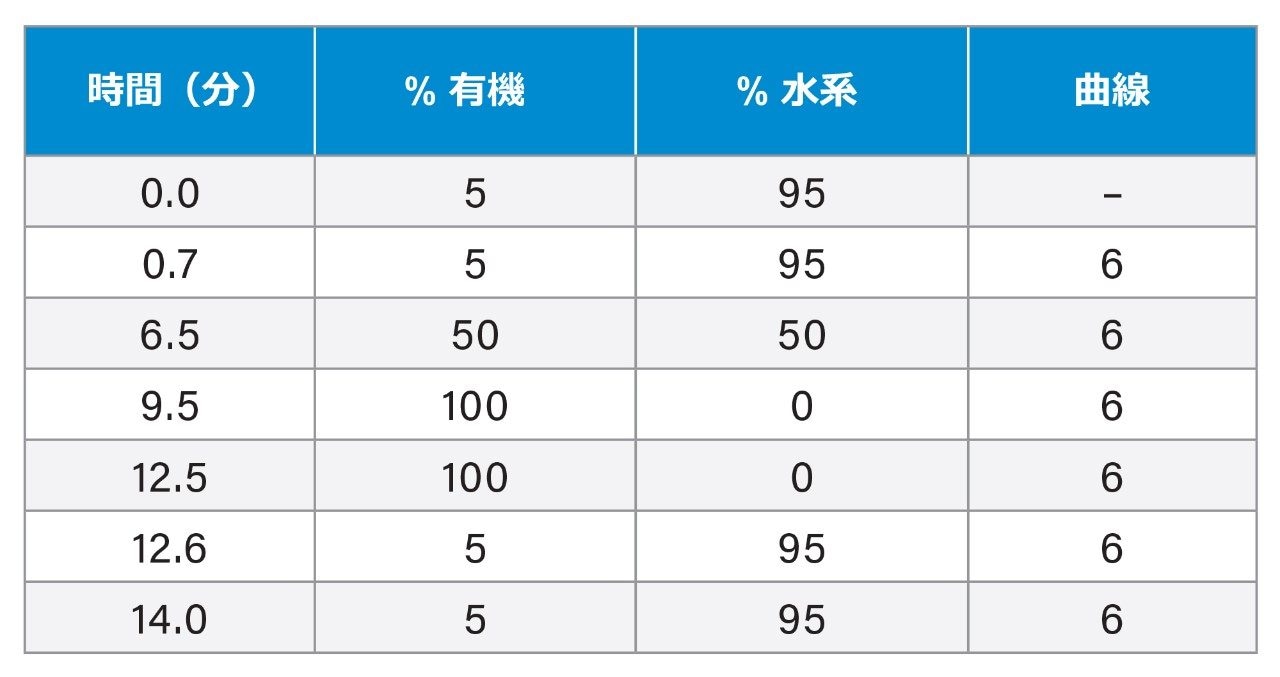 表 1. UPLC グラジエント
表 1. UPLC グラジエント
MS 条件
|
質量分析システム: |
Xevo TQ-XS |
|
イオン化モード: |
ESI+/-(極性切り替え) |
|
取り込みモード: |
マルチプルリアクションモニタリング(MRM) |
|
キャピラリー電圧: |
+0.75/-0.30 kV |
|
イオン源オフセット: |
30 V |
|
コーンガス流量: |
200 L/時間 |
|
脱溶媒温度: |
600 ℃ |
|
脱溶媒ガス流量: |
1,100 L/時間 |
|
イオン源温度: |
150 ℃ |
|
データ取り込みソフトウェア: |
MassLynx v4.2 |
|
データ解析ソフトウェア: |
定量用 waters_connect (MS Quan アプリケーション) |
最適化した MRM トランジション、コーン電圧、コリジョンエネルギーは、付録に記載しています。
結果および考察
クロマトグラフィー
モニリホルミン(保持時間 0.71 分)以外のすべての化合物は、2 つの MRM トランジションを示し、SANTE ガイドラインによるカラムのボイドボリューム(Vd 約 0.55 分)に対応する保持時間の 2 倍を上回る保持時間を示しました7。 モニリホルミンは、他のマイコトキシンとは大きく異なり、小さく非常に極性が高い酸性分子であり、その確認分析には、親水性相互作用クロマトグラフィー(HILIC)を使用した測定が望ましいと考えられます。分析種すべてにおいて、保持時間は非常に安定しており、各シーケンス内で ±0.03 分を超えるばらつきは見られませんでした。
以前に開発された UPLC 分析法を使用して、ほぼすべてのターゲット化合物について、優れたクロマトグラフィー分離が達成されました。これにより、MRM 取り込みウィンドウの最適化ができ、機能の重複を最低限に抑えることにより、デュエルタイムを最大化することができました。このクロマトグラフィーピークの分離を高感度な Xevo TQ-XS と組み合わせることで、同一の MS 分析法に 50 種の化合物を含めることが可能になりました。ターゲット化合物をスパイクした穀物抽出物の代表的なクロマトグラム 2 つを図 1 に示します。
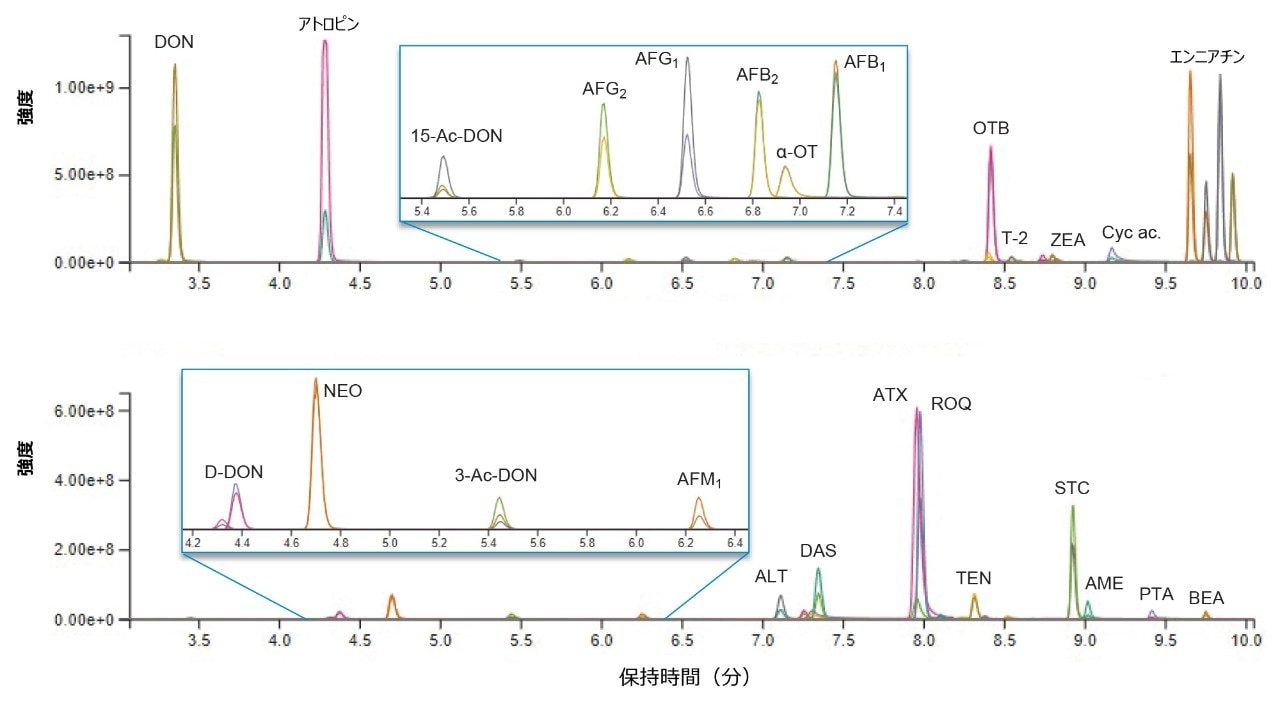 図 1. 2 種類のスパイク済み穀物サンプルにおいて試験したマイコトキシンおよび植物毒素のクロマトグラム。各ピークの特性解析を、定量イオントレースおよび定性イオントレースで行いました。
図 1. 2 種類のスパイク済み穀物サンプルにおいて試験したマイコトキシンおよび植物毒素のクロマトグラム。各ピークの特性解析を、定量イオントレースおよび定性イオントレースで行いました。
感度および定量
検出限界および定量限界に対応するクロマトグラフィーピークでは、それぞれ 3 および 10 を上回る S/N が得られました(ノイズ範囲設定:ピーク間、追加の解析なし)。
溶媒キャリブラントの分析により、極めて低い装置の検出限界(i-LOD)が達成されました。例えば、アフラトキシン B1、B2、G1、G2、M1 については、i-LOD は 0.0003 ng/mL でした。分析法の定量限界(m-LOQ)については、S/N > 10 でキャリブレーションの許容基準(R2 ≥ 0.99 および残差 ≤ 20%)に合格する最低濃度のマトリックスマッチド標準は、アフラトキシンでは 0.1 µg/kg、オクラトキシン、ステリグマトシスチン、ビューベリシン、エンニアチン、アトロピン、スコポラミン、ロケフォルチン C では ≤1 µg/kg と、低い値でした。一方、残りの化合物の m-LOQ は ≤10 µg/kg でした(ニバレノール、ゼアラレノン、ペニトレム A 以外)。表 2 の結果から、すべての 50 種の化合物についての分析法の感度および定量特性がわかります。
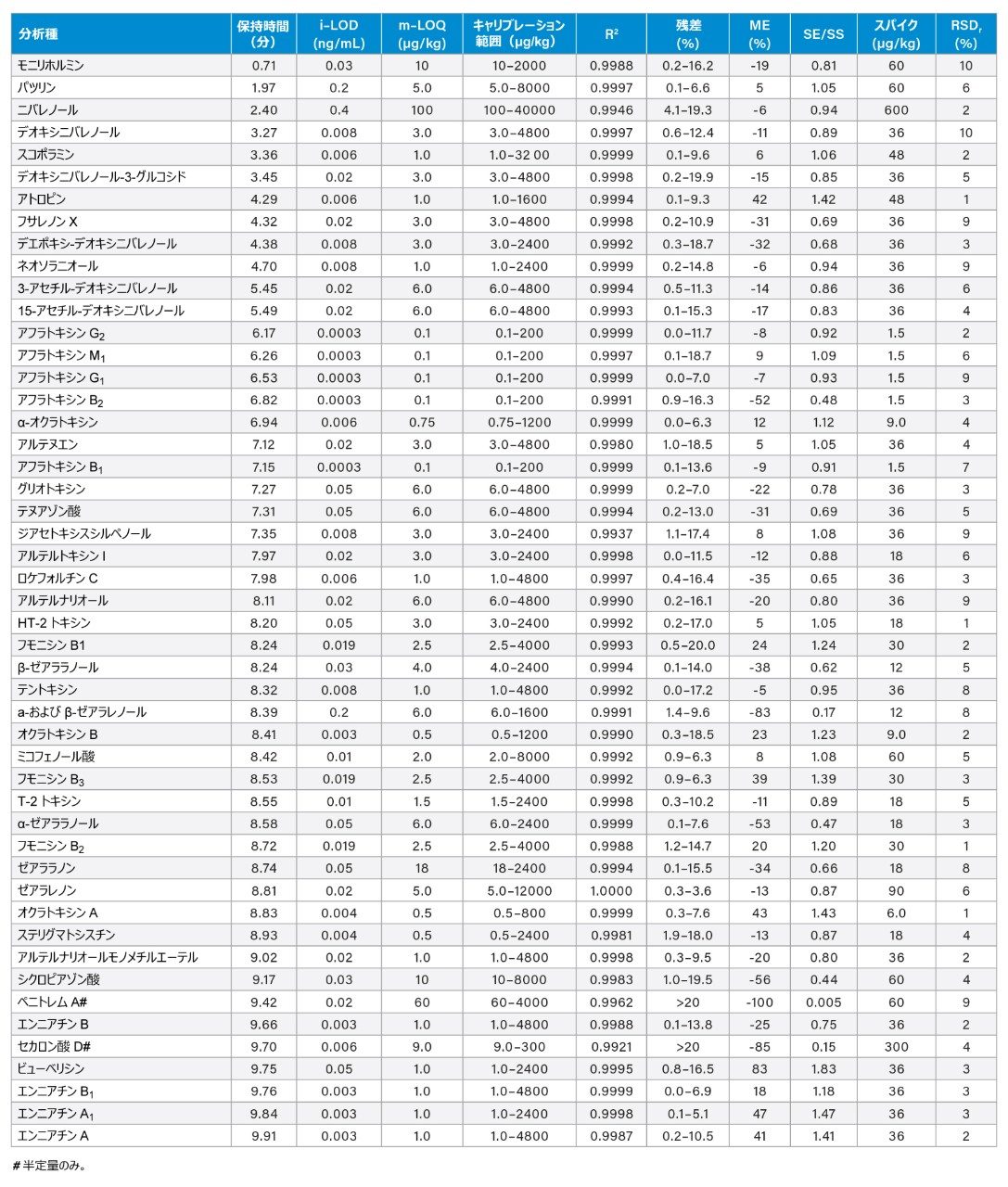 表 2. LC-MS/MS 分析法の性能評価に使用したパラメーターのサマリー。i-LOD = 装置の検出限界、m-LOQ = 分析法の定量限界、R2 = 決定係数、ME = マトリックス効果、SE/SS = シグナル増強/シグナル抑制係数、スパイク = 再現性を測定するためにサンプルにスパイクした濃度、RSDr = 再現性条件における相対標準偏差(%)。
表 2. LC-MS/MS 分析法の性能評価に使用したパラメーターのサマリー。i-LOD = 装置の検出限界、m-LOQ = 分析法の定量限界、R2 = 決定係数、ME = マトリックス効果、SE/SS = シグナル増強/シグナル抑制係数、スパイク = 再現性を測定するためにサンプルにスパイクした濃度、RSDr = 再現性条件における相対標準偏差(%)。
キャリブレーショングラフの範囲は、ほとんどの分析種で 3 桁の範囲でした。決定係数(R2)は、溶媒およびマトリックスマッチド検量線のいずれにおいても、すべてほぼ > 0.99 であり、キャリブレーション範囲は残差の割合が 20% 未満であった標準のみを使用して決定しました。キャリブレーション特性の詳細は、表 2 に記載しています。
図 2 に、m-LOQ レベルのアフラトキシン B1 およびオクラトキシン A をスパイクした混合穀物抽出物のクロマトグラムを示します。それぞれのマトリックスマッチドキャリブレーショングラフは図 3 に示します。
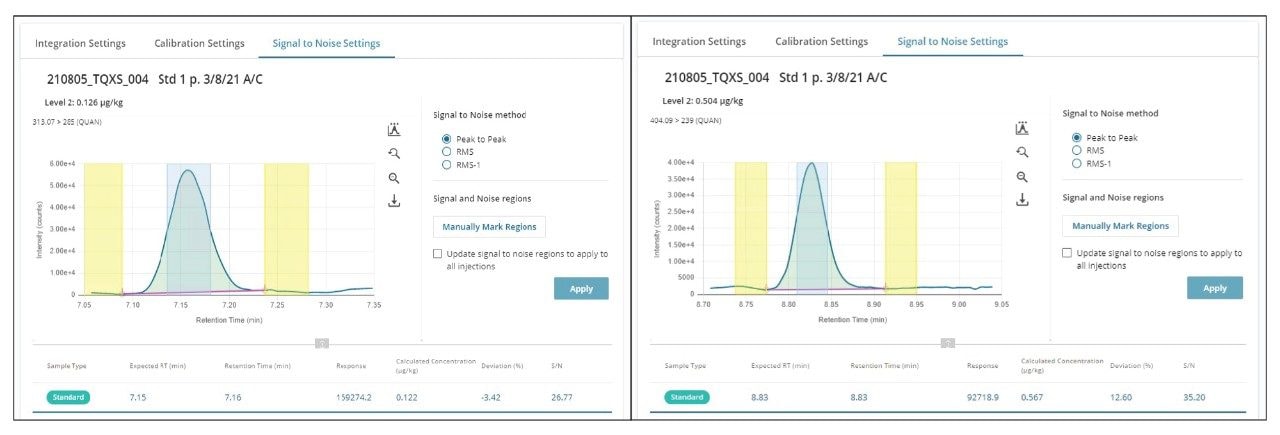 図 2. 混合穀物抽出物中のアフラトキシン B1(0.1 µg/kg)およびオクラトキシン A(0.5 µg/kg)を示す、マトリックスマッチド標準の分析のクロマトグラム。シグナル対ノイズ比の計算により、ソフトウェアのアルゴリズムによって、シグナル領域(緑色)およびノイズ領域(黄色)が自動的に同定されました。
図 2. 混合穀物抽出物中のアフラトキシン B1(0.1 µg/kg)およびオクラトキシン A(0.5 µg/kg)を示す、マトリックスマッチド標準の分析のクロマトグラム。シグナル対ノイズ比の計算により、ソフトウェアのアルゴリズムによって、シグナル領域(緑色)およびノイズ領域(黄色)が自動的に同定されました。
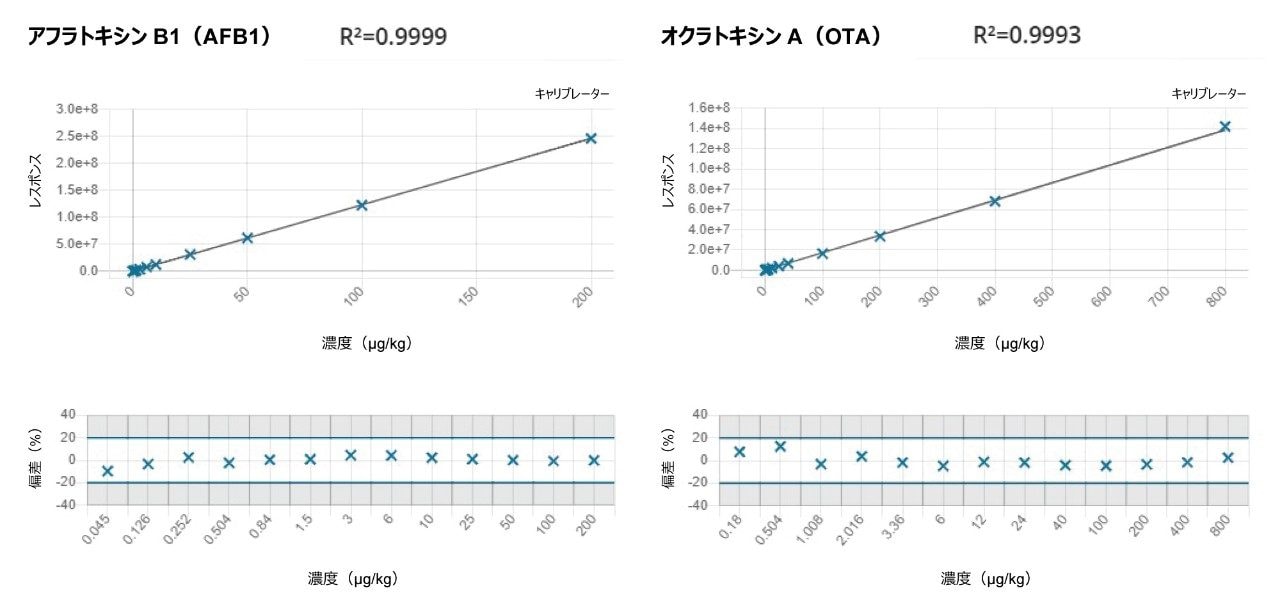 図 3. アフラトキシン B1 およびオクラトキシン A のマトリックスマッチド検量線および残差プロット
図 3. アフラトキシン B1 およびオクラトキシン A のマトリックスマッチド検量線および残差プロット
マトリックス効果
各分析種について、以下のようにマトリックス効果(%ME)を計算しました。

ここで、bM および bS はそれぞれ、マトリックスマッチド検量線および溶媒検量線の勾配です。
シグナルの増幅/抑制係数(SE/SS)を、比率 bM/bS として計算しました。
マトリックス効果の範囲は -100 ~ +83% です。シグナル増幅係数は 1.05 ~ 1.83 の範囲で、シグナル抑制係数は 0.95 ~ 0.005 の範囲でした。
観測されたマトリックス効果の程度が顕著に異なることから、マトリックスマッチドキャリブレーションを使用することが、マトリックス効果を補正し、信頼性の高い定量を達成するために必須であることが確認されました。別の方法として、13C 標識内部標準を使用することもできます5,6。
多くの場合、マトリックス共抽出物の存在が、マトリックス効果の原因となっています。複雑な食品マトリックスの場合、分析法の頑健性を向上させ、装置のメンテナンスによる装置のダウンタイムを最小限に抑え、カラムを長持ちさせるためには、有効なサンプルクリーンアップステップや抽出物の大幅な希釈を含めることが推奨されます。以前の文書8に、有効な SPE クリーンアッププロトコルの例を記載しています(製品番号:720007377EN)。
再現性および同定基準
LC-MS/MS 分析法の再現性を評価するために、コムギ、オオムギ、コメ、トウモロコシの穀粉の混合物からなる 6 つの被験サンプルに、検量線のレベル #5 の濃度と同等レベルのターゲットマイコトキシンおよび植物毒素をスパイクしました(表 2 参照)。それぞれの被験サンプルを抽出して分析しました。相対標準偏差(%RSDr)の値は、すべての分析種で ≤10% でした。いずれの場合も、スパイク済みサンプルのイオン比と保持時間は、すべての化合物について、SANTE ガイドラインに指定されている基準と一致していました7。
結論
この UPLC-MS/MS 分析法は、規制対象マイコトキシン、規制対象トロパンアルカロイド、および広範囲に及ぶ新規マイコトキシンおよびマスクドマイコトキシンの定量測定に適しています。入手可能な場合は、マトリックスマッチドキャリブレーションの代替策として 13C 標識内部標準の使用が推奨されます。
Xevo TQ-XS の究極の感度により、サンプル抽出物をかなり希釈することができ、それでも極度に低い定量限界を達成することができます。これにより、複雑なサンプル調製物の必要性が低減し、シンプルで迅速な「希釈して注入」アプローチが適用できることが実証されました。それでもなお、Oasis PRiME HLB SPE カートリッジを使用した SPE パススルーなどのクリーンアップステップを導入することで、注入前にマトリックス共抽出物の含有量を減らし、分析法および装置の頑健性を向上させることができる優れた選択肢となります。
効果的な UPLC 分離をタンデム四重極質量分析計の高感度で高選択性と組み合わせることで、分析法を更に拡張する機会が得られ、今後の必要性に応じて、より多くの分析種を含めることができます。
最後に、定量ソフトウェアに waters_connect を導入することで、データ解析の効率が最大化し、定量結果をより迅速にレビューおよび報告することができます。
参考文献
- Eskola, et al. Worldwide Contamination of Food-Crops With Mycotoxins: Validity of the Widely Cited ‘FAO Estimate’ of 25%. Crit. Rev. Food.Sci.Nutr. 60(16):2773–2789. 2020.
- Gruber-Dorninger et al.Emerging Mycotoxins: Beyond Traditionally Determined Food Contaminants.J. Agric.Food Chem. 65(33):7052–7070.2017.
- Freire and Sant’Ana.Modified Mycotoxins: An Updated Review on Their Formation, Detection, Occurrence, and Toxic Effects. Food Chem. Toxicol. 111:189–205.2018.
- de Nijs et al. Short Inventory of EU Legislation on Plant Toxins in Food. World Mycotoxin J. 9: 129–139.2017.
- Nicola Dreolin, Sara Stead.LC-MS/MS Method Development and Validation for the Quantitative Determination of Regulated Mycotoxins in Cereal Grain Flours Using Simplified Sample Preparation Conditions on Xevo TQ-XS.Waters Application Note 720006685EN, revised 2021.
- Nicola Dreolin, Janitha De-Alwis, Dimple D. Shah, Joanne Williams, Sarah Dowd, Nicole Baumgarten, Stuart Adams, Simon Hird. Evaluation of the Performance of a Simple Method for Regulated Mycotoxins in Cereals by LC-MS/MS Using an Interlaboratory Study.Waters Application Note, 720007165EN, 2021.
- SANTE 12089/2016.Guidance Document on Identification of Mycotoxins in Food and Feed.
- Nicola Dreolin, Sara Stead, Simon Hird, Timothy Jenkins. Determination of Regulated and Emerging Mycotoxins in Cereals, Nuts, Figs, and Animal Feeds Using Pass-through SPE and UPLC-MS/MS. Waters Application Note, 720007377EN, 2021.
付録
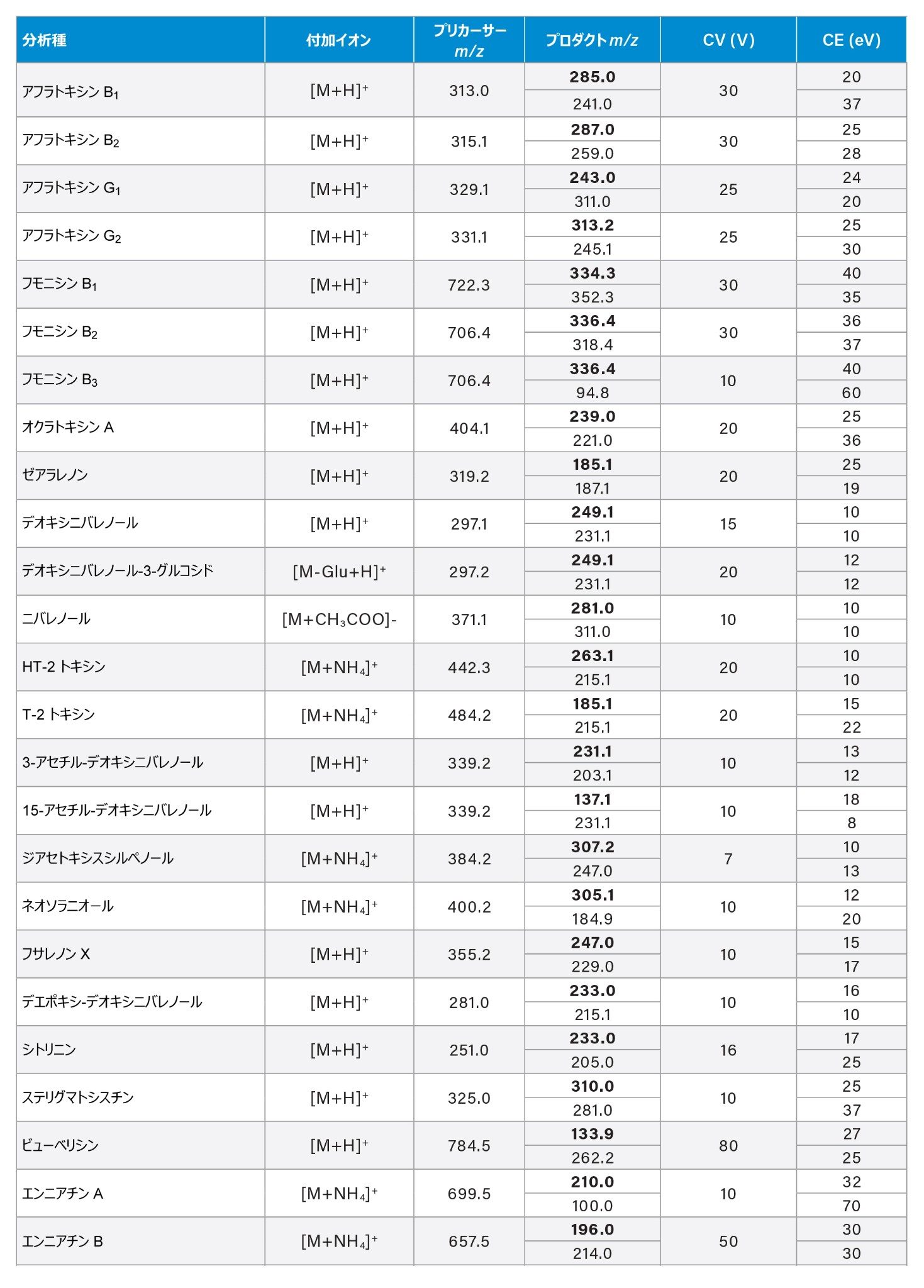
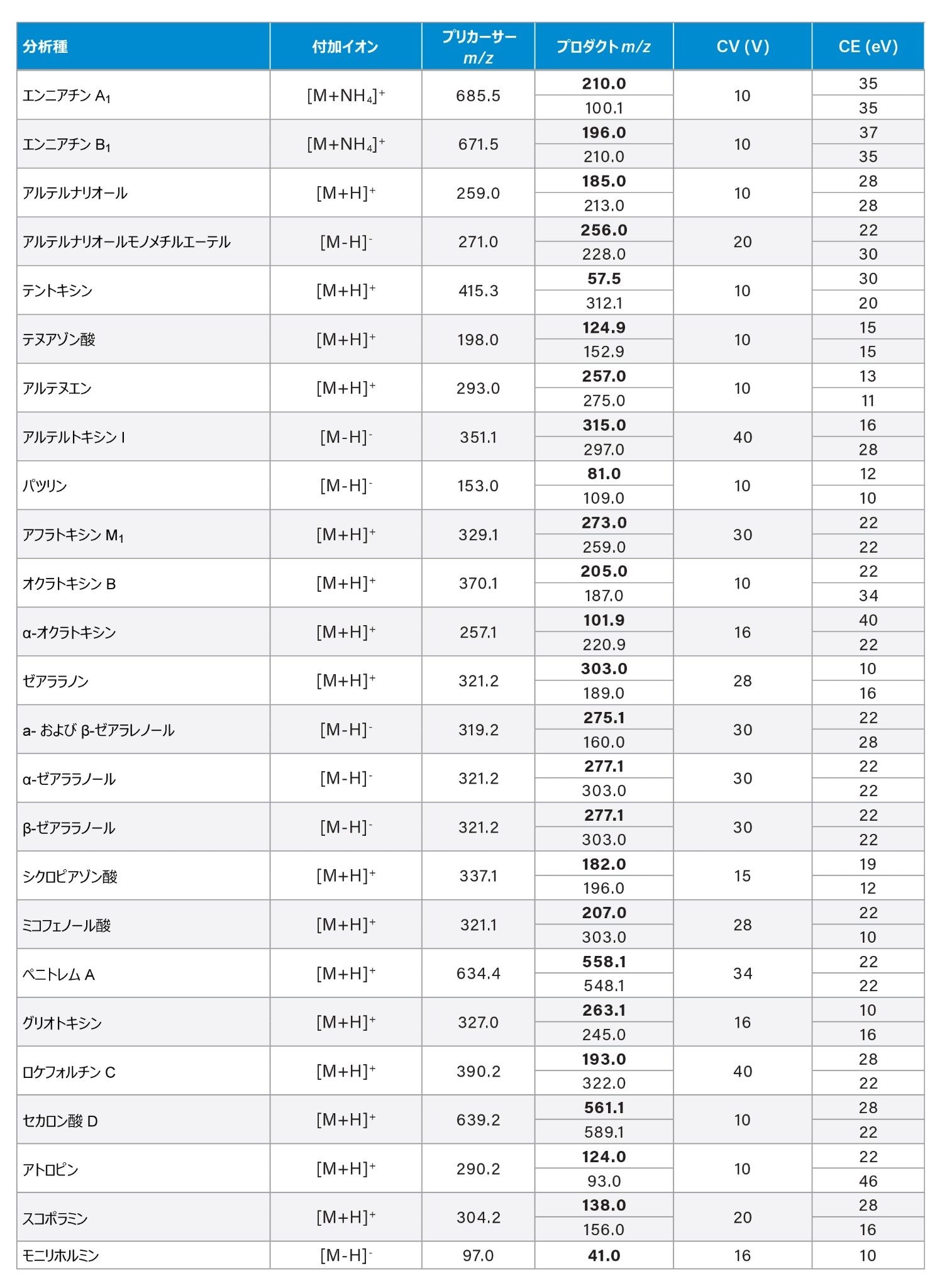 付録表 3 ターゲット分析種の最適化した MRM トランジション、サンプリングコーン電圧(CV)、コリジョンエネルギー(CE)
付録表 3 ターゲット分析種の最適化した MRM トランジション、サンプリングコーン電圧(CV)、コリジョンエネルギー(CE)
720007476JA、2021 年 12 月